尝试Autodock Vina批量分子对接
Autodock Vina批量分子对接安装软件准备配体分子和受体蛋白pdb受体蛋白pbd结构获取:配体分子pbd结构获取:预处理配体分子和受体蛋白新的改变功能快捷键合理的创建标题,有助于目录的生成如何改变文本的样式插入链接与图片如何插入一段漂亮的代码片生成一个适合你的列表创建一个表格设定内容居中、居左、居右SmartyPants创建一个自定义列表如何创建一个注脚注释也是必不可少的KaTeX数学公式
【失败】Autodock Vina批量分子对接实验记录
安装软件
AutoDock Vina下载地址
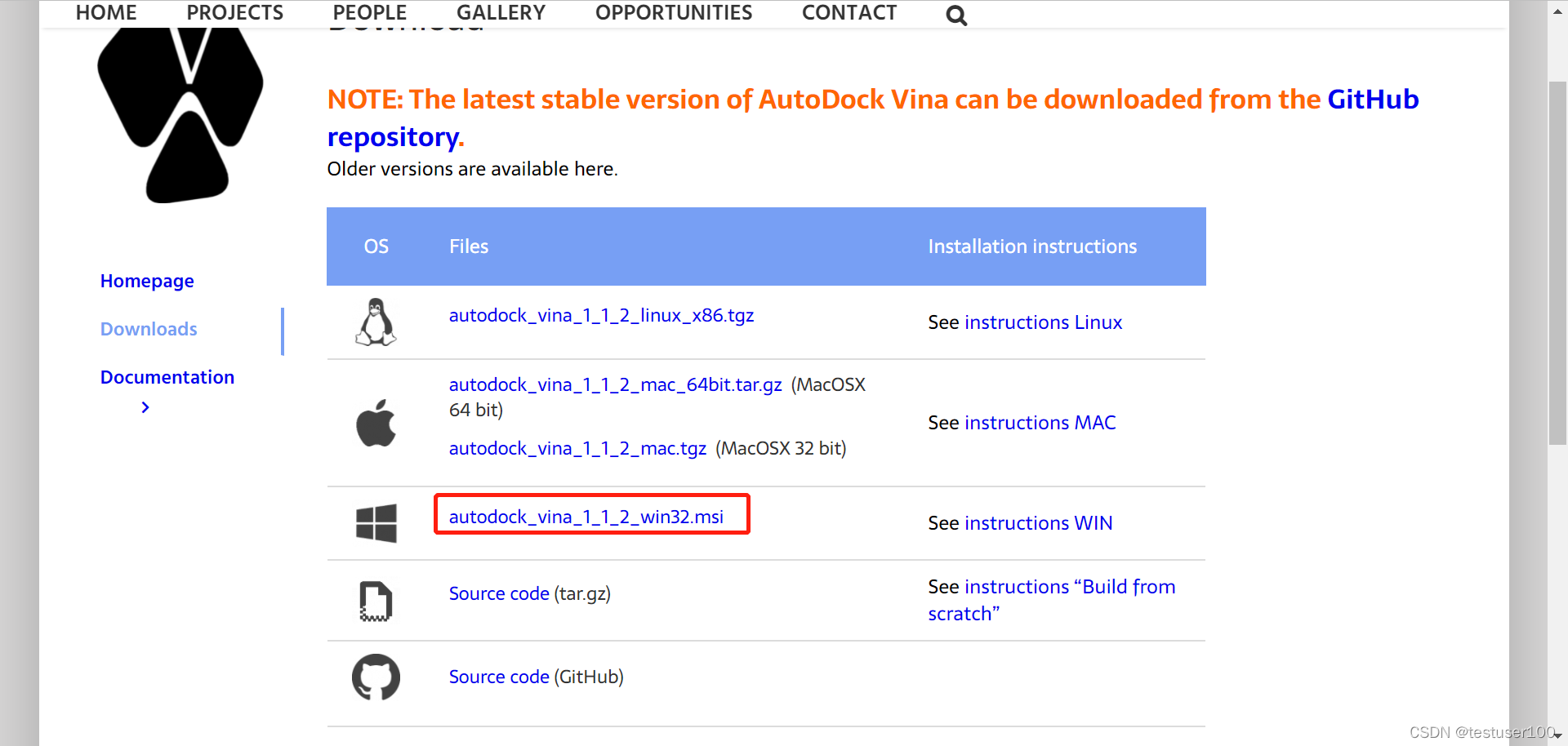
MGLTools下载地址

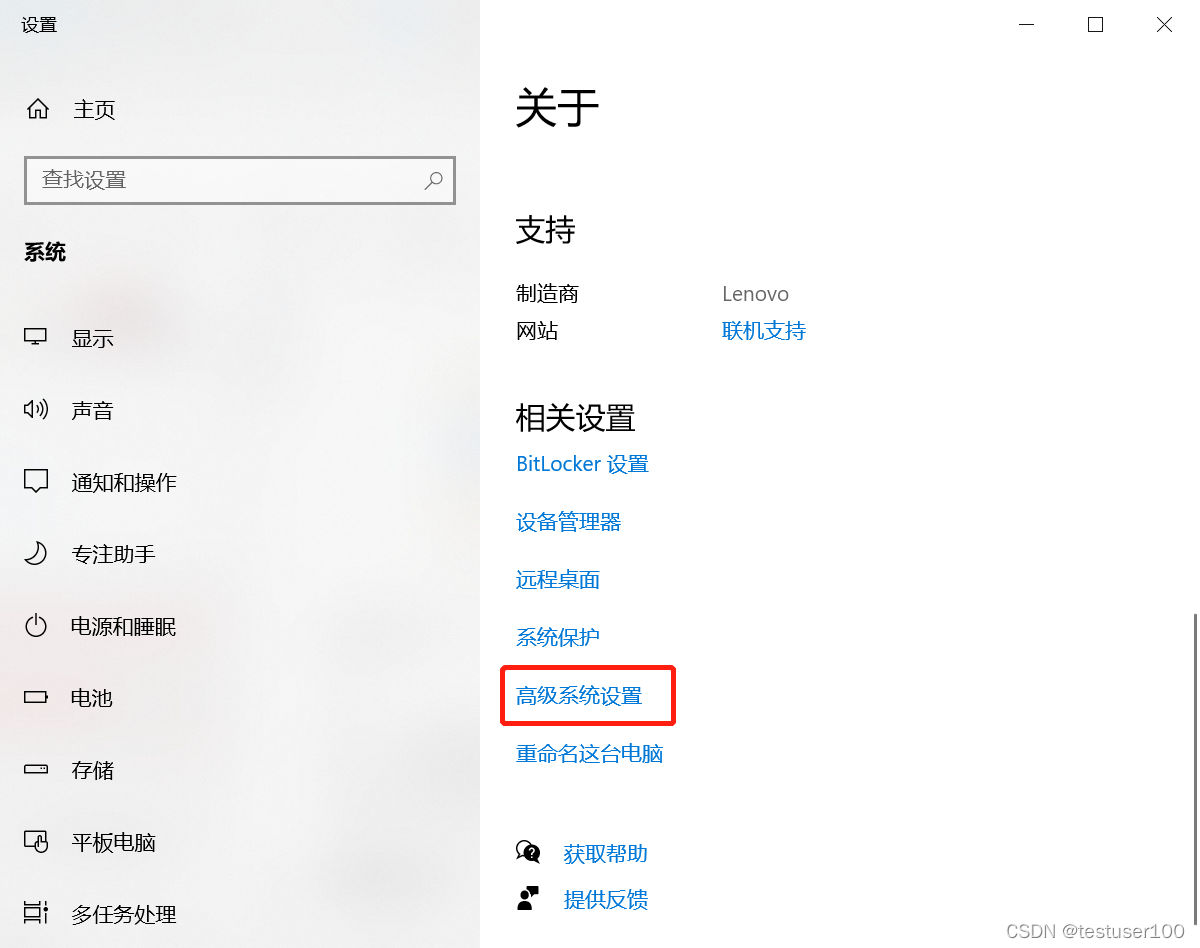
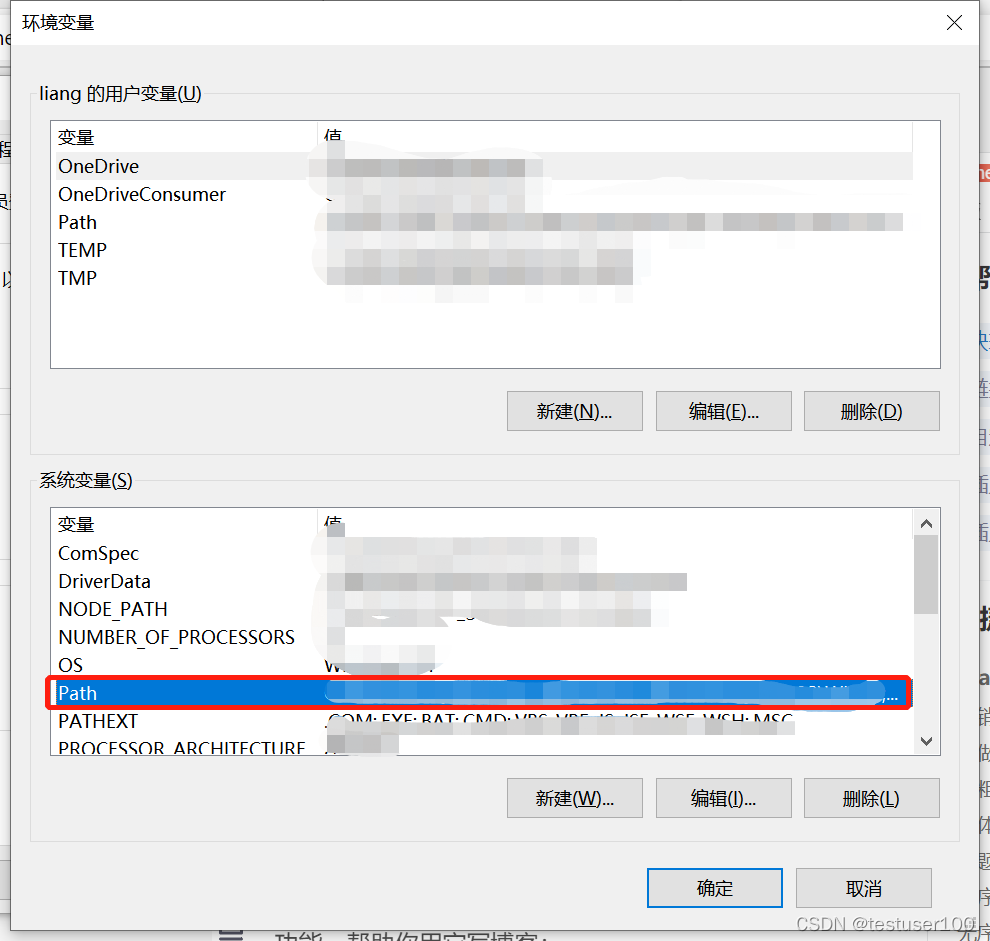
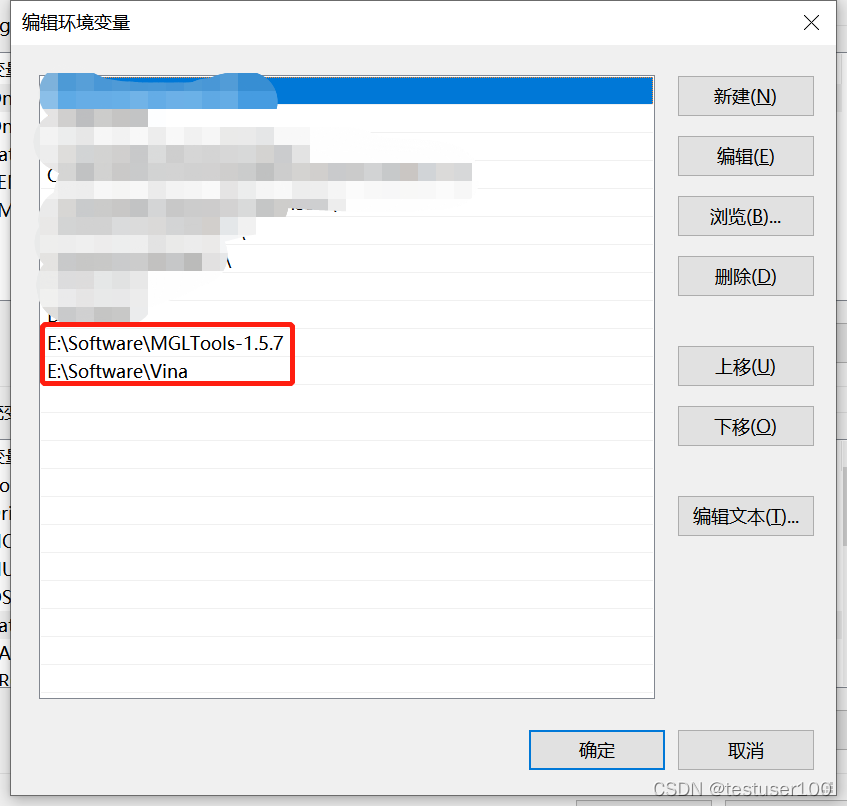
右键“此电脑” > 高级系统设置 > 环境变量 > 双击系统变量的path > 将vina和MGLTools的地址写入

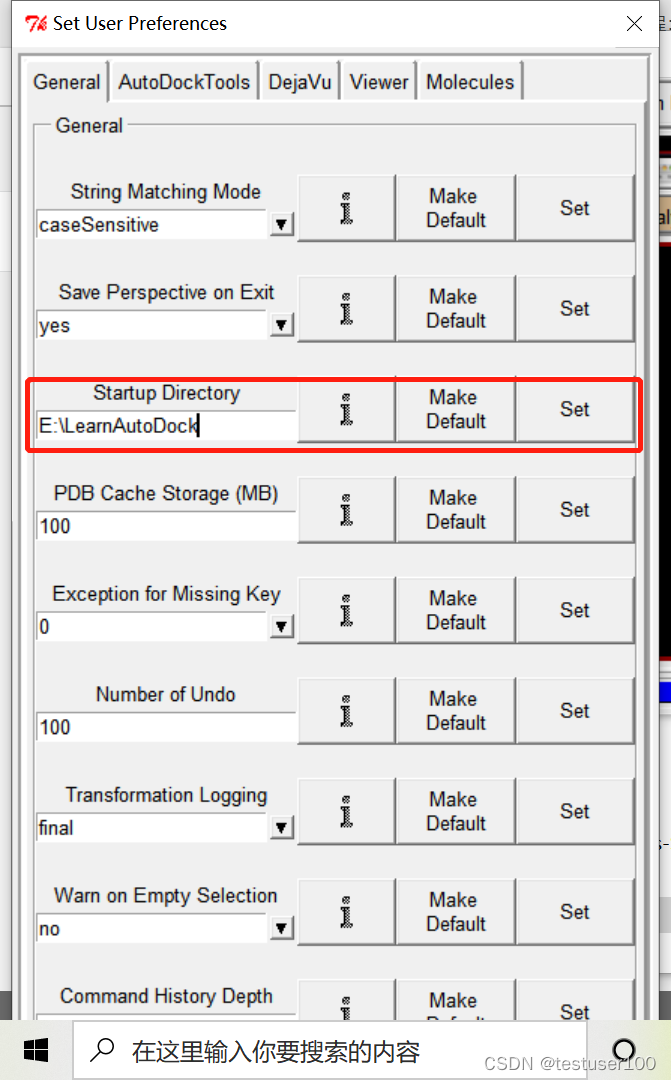
设置工作空间:打开AutoDockTools

File > Preference > set 在Start Directory设置默认的工作空间(放置蛋白分子和配体的文件夹)
open babel下载地址

准备配体分子和受体蛋白pdb
受体蛋白pbd结构获取:
在UniProt根据uniprot编号搜索得到受体蛋白的pdb文件
配体分子pbd结构获取:
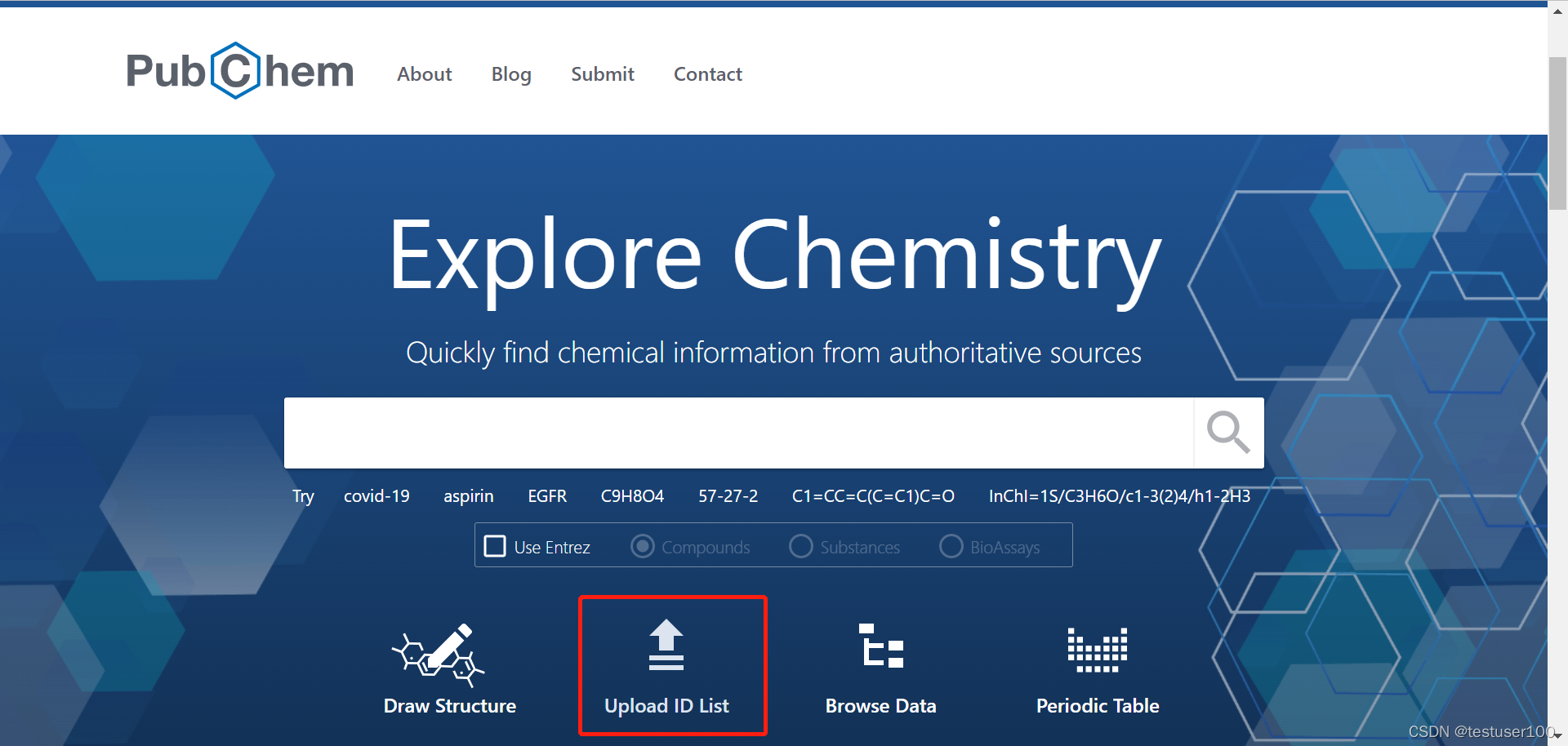
在PubChem输入ID列搜索批量的小分子。下载小分子的3D结构的sdf文件。
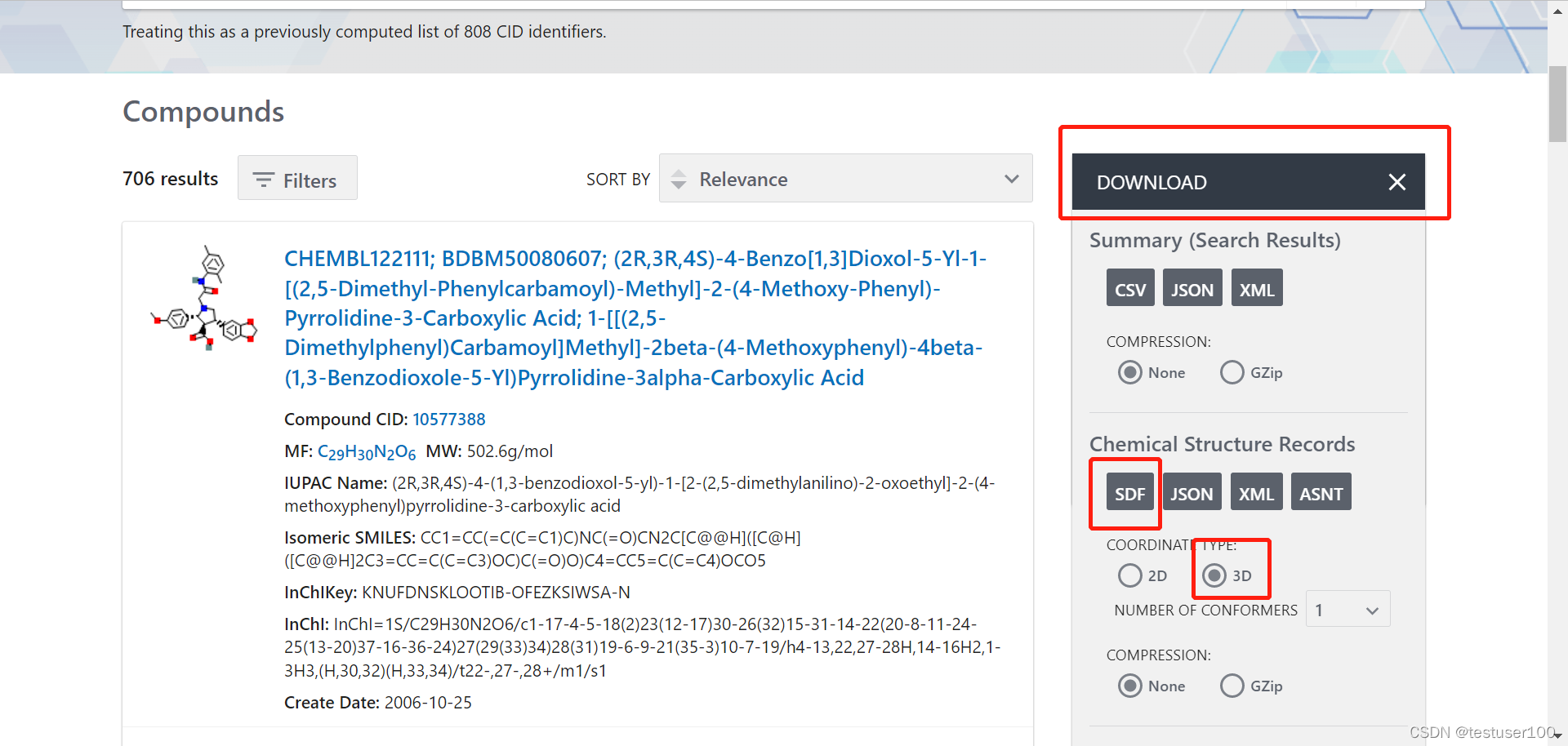
参考此链接用openbabel拆分sdf文件。
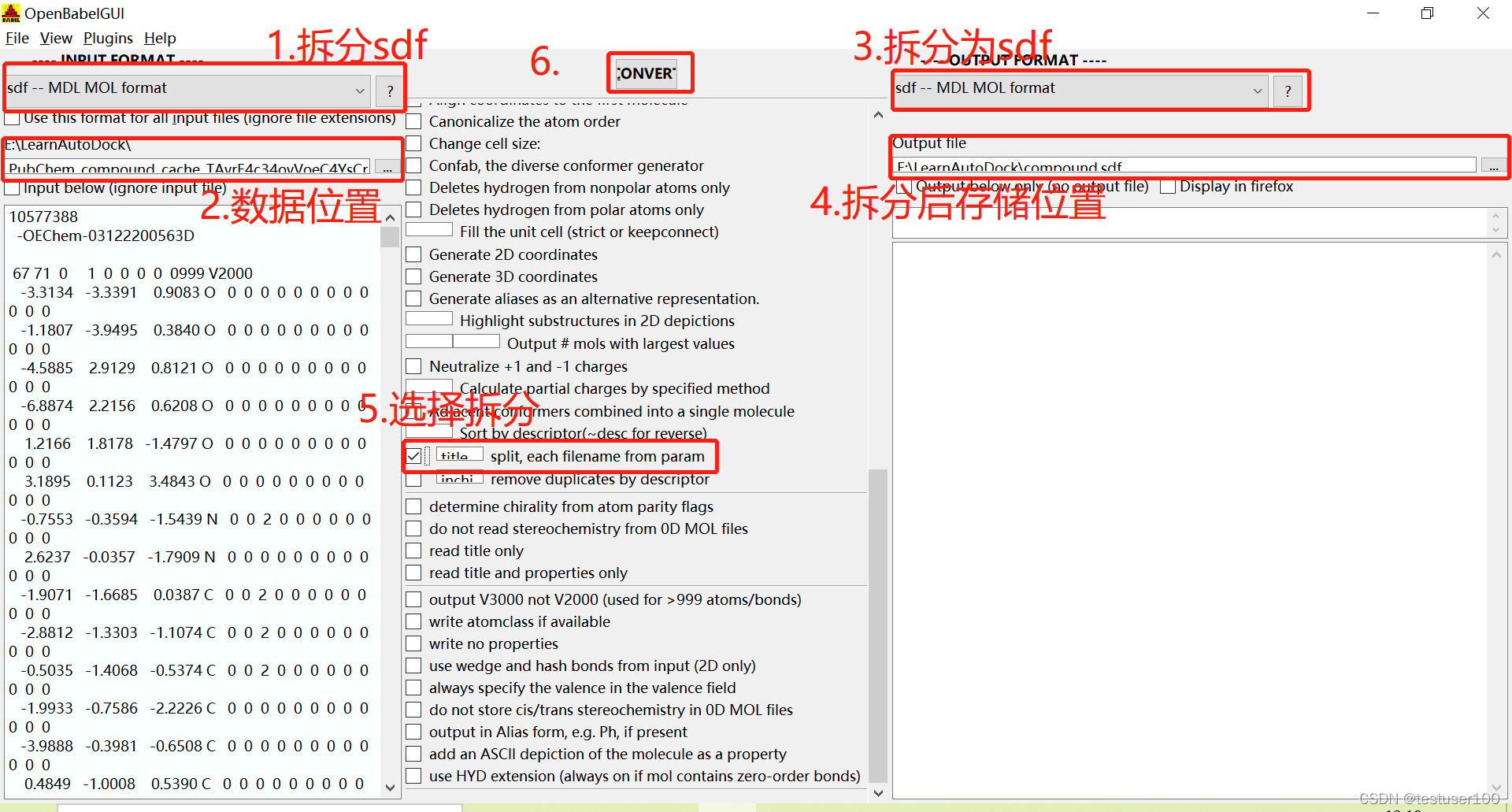
预处理配体分子和受体蛋白
用pymol打开蛋白受体,执行两个命令remove solvent(去除溶剂)和remove organic(去除小分子)即可。
安装pymol

操作蛋白分子加氢,参考link
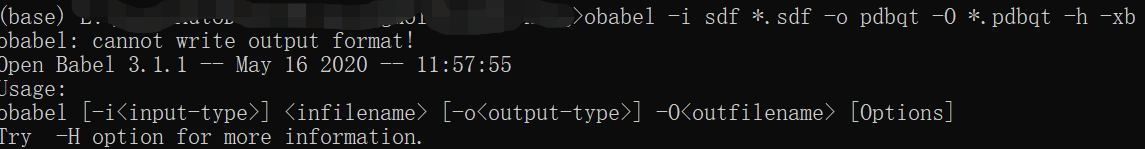
操作小分子加氢 ,但是没有成功(如图)。而且有博主说obabel直接转换为pdbqt不能使用。所以就换了Meeko方式转换。先将mk_prepare_ligand.py放在工作文件夹中,再进行run。(此处自动为小分子加氢)
obabel *.sdf -O *.pdbqt -h -xb
conda install -c rdkit rdkit
pip install meeko
mk_prepare_ligand.py
python mk_prepare_ligand.py -i 4477.sdf -o 4477.pdbqt
同时为了能够批量转换pdbqt,创建getpdbqt.txt文件,写入以下代码。保存后将后缀名改为bat。
@echo
for %%f in (*.sdf) do (
python mk_prepare_ligand.py -i %%f -o ligand_%%~nf.pdbqt
timeout 3)
exit
对接盒子(Grid Box)的设置
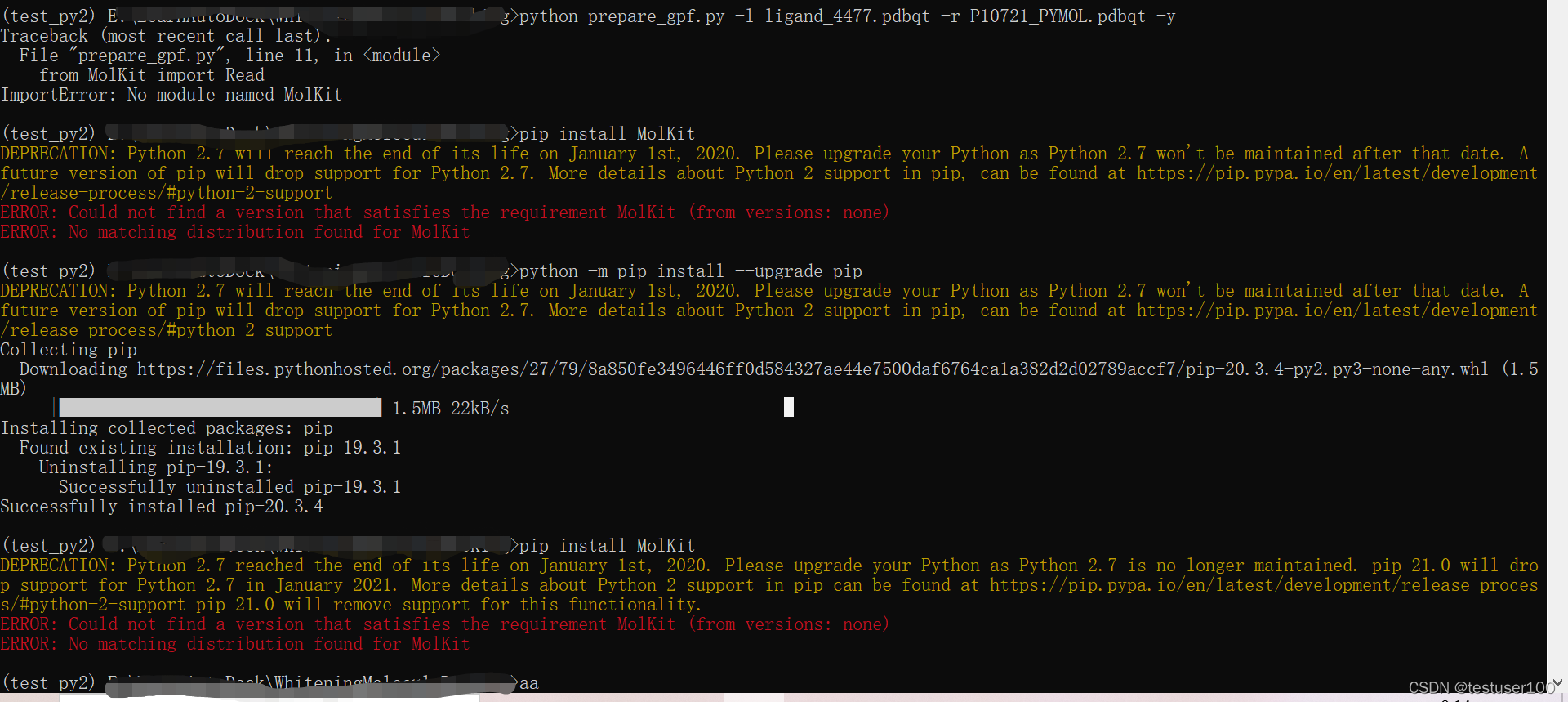
下载prepare_gpf后,用以下命令进行。但是这个文件是python2的,不适用Python3。我尝试创建Python2的虚拟环境运行,但是安装包的时候出现了错误,无法进行运行。
python prepare_gpf.py -l ligand_4477.pdbqt -r P10721_PYMOL.pdbqt -y
所以我参考利用pyMOL插件GetBox Plugin确定分子对接口袋 下载GetBox计算受体蛋白的对接位置。但是可能是因为我是直接在uniprot数据库搜索蛋白编号得到的蛋白,无法计算出对接位置。
然后我就把蛋白分子导入mgltools用grid box,手动将蛋白分子全包围了。参考分子对接教程 | (6) AutoDock对接操作与对接结果解读

结果分析
参考win10中使用AutoDock Vina进行批量对接的批处理程序进行批量处理分子对接,但是这样全包围分子对接,一个配体的运行时间几乎要2小时,代价太高。所以正在想其他方法(或者外包,毕竟我的专业和这个完全打不着)。
conf.txt
receptor = P10721_PYMOL.pdbqt
center_x= -7.989
center_y= 0.833
center_z= -4.906
size_x = 126
size_y = 126
size_z = 126
num_modes = 9
dock.bat
@echo
for %%f in (ligand_*.pdbqt) do (
echo Processing ligand %%f
if not exist "%%~nf" mkdir "%%~nf"
vina --config conf.txt --ligand %%f --out "%%~nf"/out.pdbqt --log "%%~nf"/log.txt
timeout 3)
exit
用这两个文件跑了很长时间得到一个结果(如下)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -8.1 0.000 0.000
2 -8.0 2.517 7.118
3 -7.7 2.433 4.967
4 -7.6 49.089 50.925
5 -7.6 4.017 8.350
6 -7.4 38.055 41.525
7 -7.4 18.621 21.388
8 -7.4 16.409 19.542
9 -7.4 37.367 40.743
Writing output ... done.
搜索后得到结果说明:
查看打分列表,了解各个构象的打分分布情况,比较各个化合物的打分差异。
RMSD:表示各构象相对于最佳构象(Pose 1)的重原子(除氢原子外的其他原子)空间位置差异程度。
RMSD_ub:RMSD upper bound,即构象间原子严格对应的RMSD(忽略对称性),常用于非对称分子;
RMSD_lb:RMSD lower bound,即构象间原子最佳RMSD(考虑对称性),常用于对称分子。
注意:在普通分析中,大可不必考虑该RMSD。
对接软件的打分函数给出来的一个参数。这个参数是希望用来表征配体与蛋白质口袋的结合强弱。可以认为一定程度上是为了类比delta G。
rmsd:root-mean-square deviation, 在vina里用于表明各个对接位置之间的差异。因为是以第一个位置作为参照,所以第一个位置的rmsd是0.
通常选用一个对接软件,及确定参数设置,需要做一步方法检验。常用的方法就是你对一些晶体结构中的配体用你的对接软件和参数方法做
对接,如果你对接出来的位置跟晶体结构中配体的位置的rmsd在2A范围内,基本认为你的对接软件及方法有一定的可靠性。希望有帮助
怎么确定这个affinity多大合适,完全与取决于你的体系和你想实现的目标。不同的体系都是不一样。从delta G的角度看,只要delta G小于零,
反应都是自发进行。但是作为一个药物设计,当然不能仅仅能结合就够。通常来说,绝对值越高越高。当然最后还是那句话,出发前,先要
知道自己的目的和参照,才能知道自己是否达到目标。

为开发者提供学习成长、分享交流、生态实践、资源工具等服务,帮助开发者快速成长。
更多推荐



 10
10 1
1- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)