下载NCBI的SRA数据 详细教程
SRA(Sequence ReadArchive)数据库是NCBI(National Center for Biotechnology Information)旗下用于存储高通量测序数据的子库。来自世界各地研究的测序数据在此处都可以免费下载,本文就聊聊如何下载SRA数据库对应的测序数据。1. SRA基本框架下载数据之前,咱们可以先聊聊SRA数据库的一些基础知识。SRA数据库的组织框架是基于 STU
SRA(Sequence ReadArchive)数据库是NCBI(National Center for Biotechnology Information)旗下用于存储高通量测序数据的子库。来自世界各地研究的测序数据在此处都可以免费下载,本文就聊聊如何下载SRA数据库对应的测序数据。
1. SRA基本框架
下载数据之前,咱们可以先聊聊SRA数据库的一些基础知识。
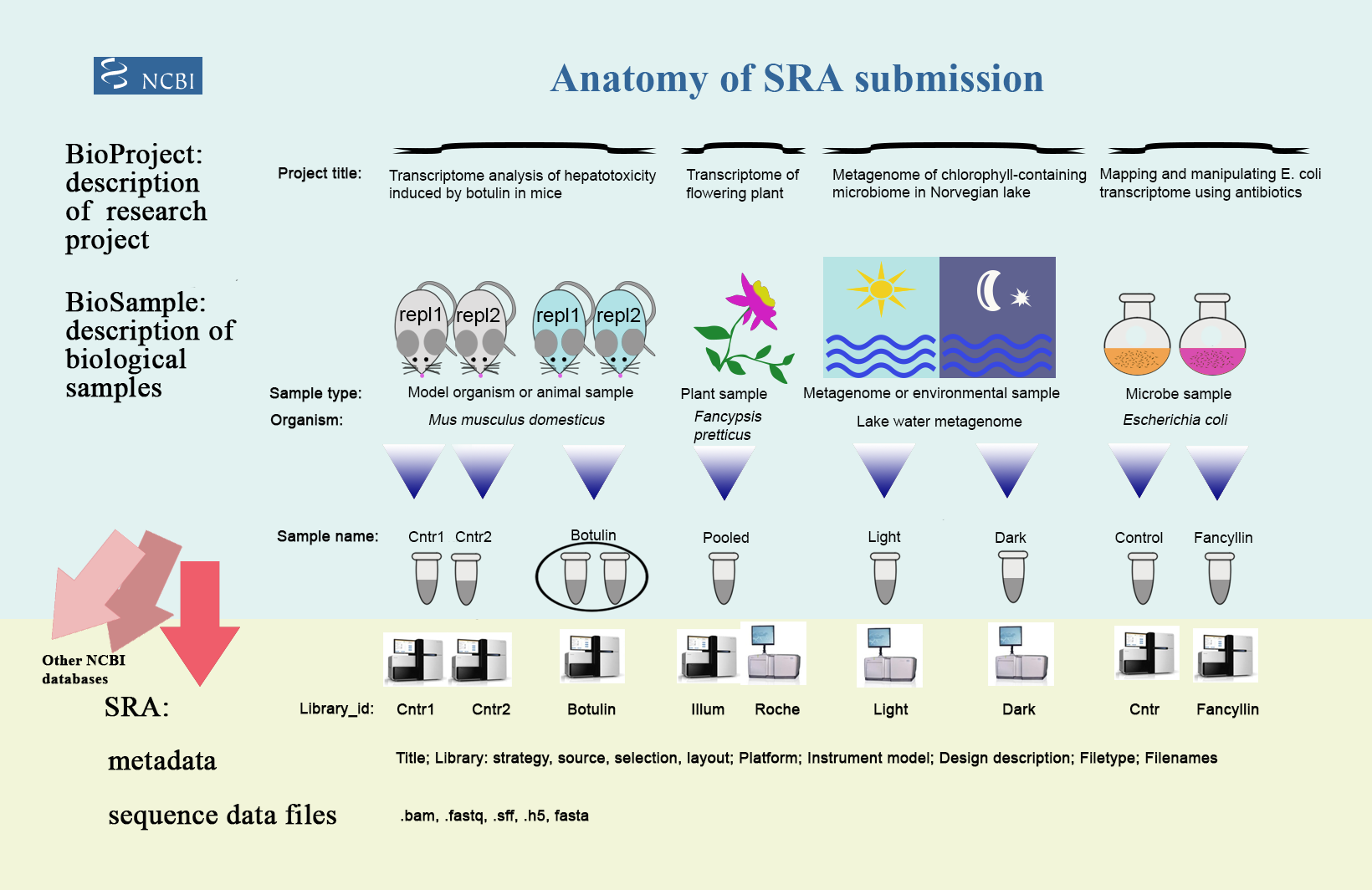
SRA数据库的组织框架是基于 STUDY, SAMPLE, EXPERIMENT, RUN 四个概念构建的。
- STUDY :研究课题/研究项目, 和NCBI的子库
[BioProject](https://www.ncbi.nlm.nih.gov/bioproject/)数据库里面的项目相关联 , 检索号(accession number) 通常以前缀SRP,DRP,ERP开头(例如SRP000544) - SAMPLE :样本信息, 和NCBI的子库
[BioSample](https://www.ncbi.nlm.nih.gov/biosample/)数据库里面的样本相关联,检索号(accession number) 通常以前缀SRS,DRS,ERS开头(例如SRS001487) - EXPERIMENT : 实验信息, 一次实验包含了一个或多个样本进行一个多多个RUN的测序。检索号(accession number) 通常以前缀
SRX,DRX,ERX开头 - RUN:RUN通俗理解就是测序仪运行一次产生的测序数据,是 SRA 里面最小的概念,该编号通常直接链接到对应某一个/对fastq下机的文件。检索号 (accession number) 通常以前缀
SRR,DRR,ERR开头。
说了这么多是不是依然傻傻分不清SRP,DRP,ERP等,其实编号的含义很简单:
第一个字母:表示样本最初被上传到的源数据库,NCBI会同步EBI和DDBJ的数据,同步后会保留源数据的来源信息。
S – NCBI’s SRA database
E – EBI’s database
D – DDBJ database
第二个字母:固定为"R",代表Read
第三个字母:数据的类型,可以是项目、样本、实验或RUN
R – Run
X – Experiment
S – Sample
P – Project / study
2. SRA数据下载
SRA数据下载有多种方式,这里我列了常用的两种方式:1)通过官网提供的 SRA-Toolkit 工具进行下载 2)直接wget/curl下载。
2.1. SRA-Toolkit 工具进行下载:
这种方式自然要先安装 SRA-Toolkit,可以直接下载对应的二进制包,解压即可使用。
对应的下载链接为https://github.com/ncbi/sra-tools/wiki/02.-Installing-SRA-Toolkit
#Ubuntu
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
#CentOS
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-centos_linux64.tar.gz
#Mac OS X
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-mac64.tar.gz
#解压 tar.gz名字以实际文件名为准
tar -xzvf sratoolkit.*.tar.gz
下载解压后,可以看到文件夹bin/目录下面有一堆二进制运行的组件,其中 prefetch,srapath , fastq-dump都是常用的工具集。
此处下载使用prefetch 工具,例如下载千人基因组项目的数据ERR009357。
prefetch ERR009357
也可以提供RUN的SRR/DRR/ERR检索号列表, 例如此处我们下载SRR_Acc_List.txt文件内的所有ERR数据:
$ cat SRR_Acc_List.txt
ERR003133
ERR003134
ERR003135
ERR003136
ERR003137
$ nohup prefetch -O . $(<SRR_Acc_List.txt) & # 此处的 nohup + & 用于放入后台下载,避免关闭终端导致下载中断
2.2. wget/curl 下载:
使用wget/curl下载需要事先知道对应sra数据的下载链接,一种方式可以通过以下链接:
https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=SRR编号 获取,其中SRR编号替换为你需要下载的RUN检索号即可,例如打开https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=ERR009357 即可查看 ERR009357对应的详细信息。其中 Data access 页面即可看到对应的下载链接。其中,Metadata, Reads 页面可以查看样本实验信息。

复制下载链接,使用wget/curl下载即可。
wget https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/ERR009357/ERR009357.4
嫌麻烦的话也可以通过 srapath 获取对应下载链接:
#srapath获取ERR009357的下载链接
$ srapath ERR009357
https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/ERR009357/ERR009357.4
#wget下载
$ wget https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/ERR009357/ERR009357.4
#curl 下载
$ curl https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/ERR009357/ERR009357.4
3. SRA数据转化为fastq文件
下载完 sra 文件,我们往往需要先将 sra 的文件转化为 fastq 文件才能用于后续分析。此处也是使用前面安装的 SRA-Toolkit 中的fastq-dump工具。
一行命令即可解决:
#SE数据
fastq-dump SRR15671203 -O ./
#PE数据
fastq-dump ERR009357.4 --split-3 -O ./
参考链接:
https://www.ncbi.nlm.nih.gov/sra/docs/submitmeta/
https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc

为开发者提供学习成长、分享交流、生态实践、资源工具等服务,帮助开发者快速成长。
更多推荐


 9
9 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)