RepeatMasker(一)——安装及使用
RepeatMasker安装及使用文章目录RepeatMasker安装及使用一、安装1. RMBlast序列搜索引擎2.TRF搜索串联重复序列3. repeatmasker下载4. Repbase数据库5. 配置repeatmasker依赖关系二、使用1.参数说明2.输入3. 使用RepeatMasker三、结果1. masked文件2. out文件3. out.gff文件(和.out文件完全一样
·
RepeatMasker安装及使用
文章目录
一、安装
1. RMBlast序列搜索引擎
# 下载RMBlast源码包并编辑
cd /share2/pub/yangjy/yangjy/softs/
wget ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/2.6.0/ncbi-blast-2.6.0+-src.tar.gz
wget http://www.repeatmasker.org/isb-2.6.0+-changes-vers2.patch.gz
tar zxvf ncbi-blast-2.6.0+-src.tar.gz
gunzip isb-2.6.0+-changes-vers2.patch.gz
cd ncbi-blast-2.6.0+-src
patch -p1 < ../isb-2.6.0+-changes-vers2.patch
cd c++
./configure --with-mt --prefix=/share2/pub/yangjy/yangjy/softs/rmblast --without-debug
make
# 安装程序及库至系统目录,有报误,但我们需要的rmblastn已经可以正常使用了
sudo make install # Makefile:40: recipe for target 'install-toolkit' failed
# 测试程序是否安装成功
/share2/pub/yangjy/yangjy/softs/rmblastn -h
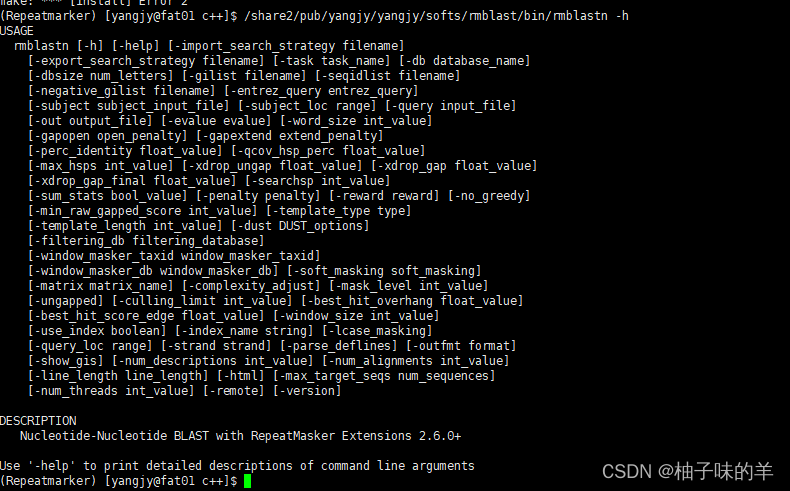
2.TRF搜索串联重复序列
cd /share2/pub/yangjy/yangjy/softs/
wget http://tandem.bu.edu/trf/downloads/trf409.legacylinux64
cp trf409.legacylinux64 ./trf
# 测试有帮助信息即可用
trf
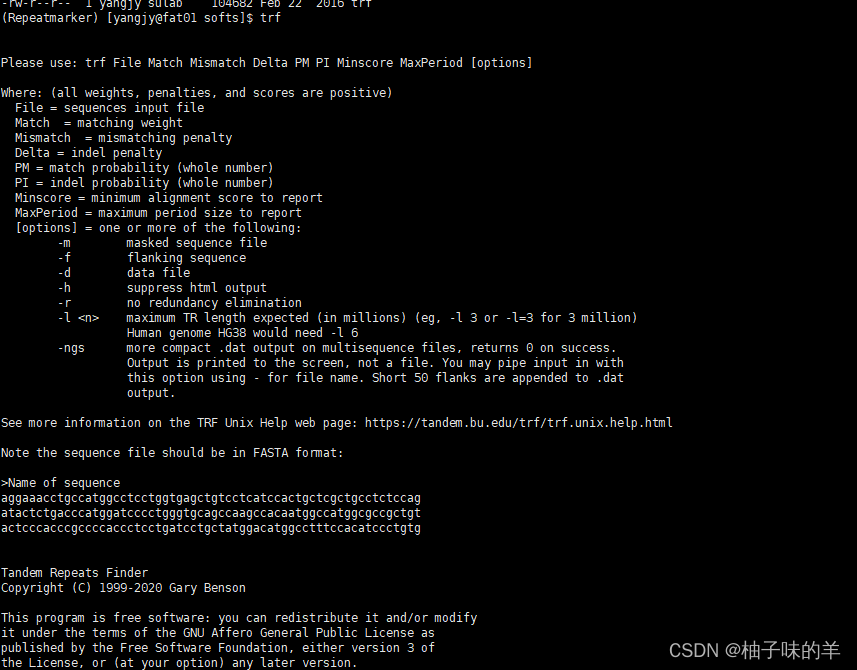
3. repeatmasker下载
cd /share2/pub/yangjy/yangjy/softs/
wget -c http://www.repeatmasker.org/RepeatMasker-open-4-0-7.tar.gz
tar xvzf RepeatMasker-open-4-0-7.tar.gz
4. Repbase数据库
我在本地下载后上传至服务器
mv RepBaseRepeatMaskerEdition-20170127.tar.gz RepeatMasker/
cd RepeatMasker/
tar xvzf RepBaseRepeatMaskerEdition-20170127.tar.gz
5. 配置repeatmasker依赖关系
# 默认perl, repatmasker, trf安装位置正确的就一路回车,搜索引挚选择2 RMBlast,输入RMBlast安装目录/share2/pub/yangjy/yangjy/softs/rmblast/bin,再选5 done完成
./configure

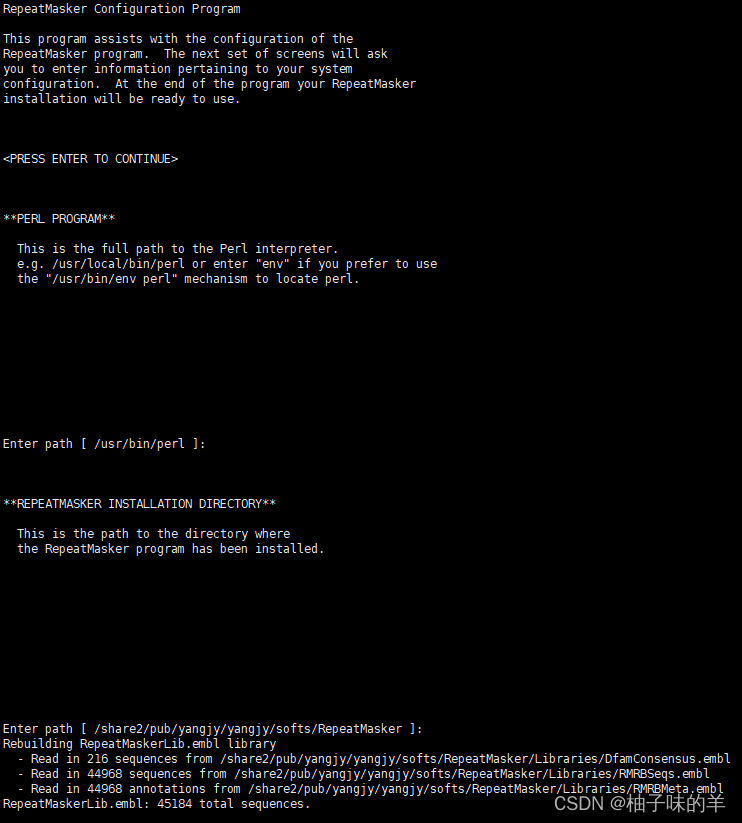
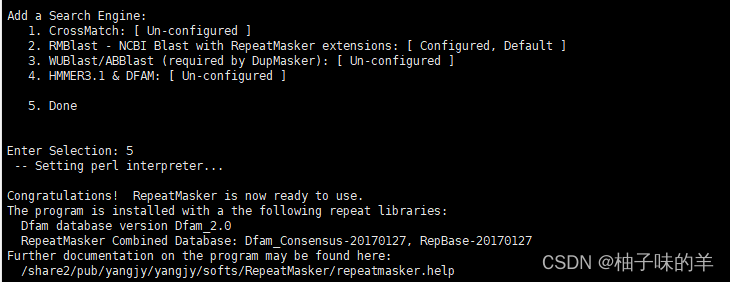
所有的位置都让软件自己识别,最后的RMBlast填写自己安装的位置。将RepeatMasker加入全局变量
#添加至全局环境变量(在bashrc中添加)
#for repeatmasker 2022.4.24
export PATH="/share2/pub/yangjy/yangjy/softs/RepeatMasker:"$PATH

试运行: RepeatMasker -h显示结果如下:
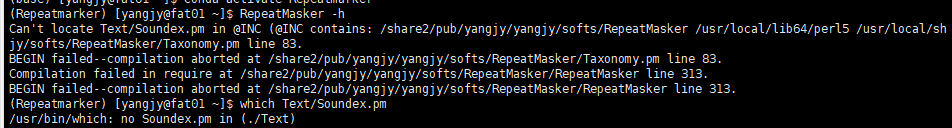
是Perl出了问题,因为Perl没有安装Text/Soundex这个module,但是不想找师兄让他在服务器上的公用目录安装,在GitHub上找到了答案,RepeatMasker 作者说现在已经不依赖Text/Soundex了,我注释了这一行(83行)之后可以正常运行
二、使用
1.参数说明
2.输入
输入是fasta格式
- 如果是下载的数据是单端测序fastq格式(只有一个fastq文件)可以使用以下方式转为fasta格式
awk '{if(NR%4 == 1){print ">" substr($0, 2)}}{if(NR%4 == 2){print}}' /share2/pub/yangjy/yangjy/rna-seq-data/GSE115828/tmp/$i.fastq > /share2/pub/yangjy/yangjy/rna-seq-data/GSE115828/fasta/$i.fasta
- 如果下载数据是双端测序fastq格式(有两个fastq文件),可以先将两个fastq文件merge后再转为fasta
cd /share2/pub/yangjy/yangjy/rna-seq-data/GSE115828/fastq_data/
f1='_1'
f2='_2'
for i in 'SRR7460825' 'SRR7460826' 'SRR7460827' 'SRR7460828' 'SRR7460829' 'SRR7460830' 'SRR7460831' 'SRR7460832' 'SRR7460833' 'SRR7460834' 'SRR7460835' 'SRR7460836' 'SRR7460837'
do
tmp_1=$i$f1
tmp_2=$i$f2
cat /share2/pub/yangjy/yangjy/rna-seq-data/GSE115828/fastq_data/$tmp_1.fastq /share2/pub/yangjy/yangjy/rna-seq-data/GSE115828/fastq_data/$tmp_2.fastq > /share2/pub/yangjy/yangjy/rna-seq-data/GSE115828/tmp/$i.fastq
awk '{if(NR%4 == 1){print ">" substr($0, 2)}}{if(NR%4 == 2){print}}' /share2/pub/yangjy/yangjy/rna-seq-data/GSE115828/tmp/$i.fastq > /share2/pub/yangjy/yangjy/rna-seq-data/GSE115828/fasta/$i.fasta
rm /share2/pub/yangjy/yangjy/rna-seq-data/GSE115828/tmp/$i.fastq
done
3. 使用RepeatMasker
RepeatMasker -pa 4 -species human -poly -a -dir /share2/pub/yangjy/yangjy/rna-seq-data/GSE146887/fasta/repeat /share2/pub/yangjy/yangjy/rna-seq-data/GSE146887/fasta/SRR11296675.fasta
注:-a 会生成一个align文件,显示是v还是i
三、结果
原来的运行代码如下,共生成了7个文件
RepeatMasker -pa 4 -species human -poly -html -gff -a -dir /share2/pub/yangjy/yangjy/rna-seq-data/GSE146887/fasta/repeat /share2/pub/yangjy/yangjy/rna-seq-data/GSE146887/fasta/SRR11296675.fasta
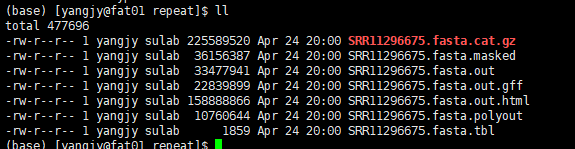
1. masked文件
可以看到发生重复序列的位置,其中直接使用N将重复序列掩盖,可以在运行的过程中加上参数-xsmall 将重复区域全部转化为小写字母,不需要N掩盖
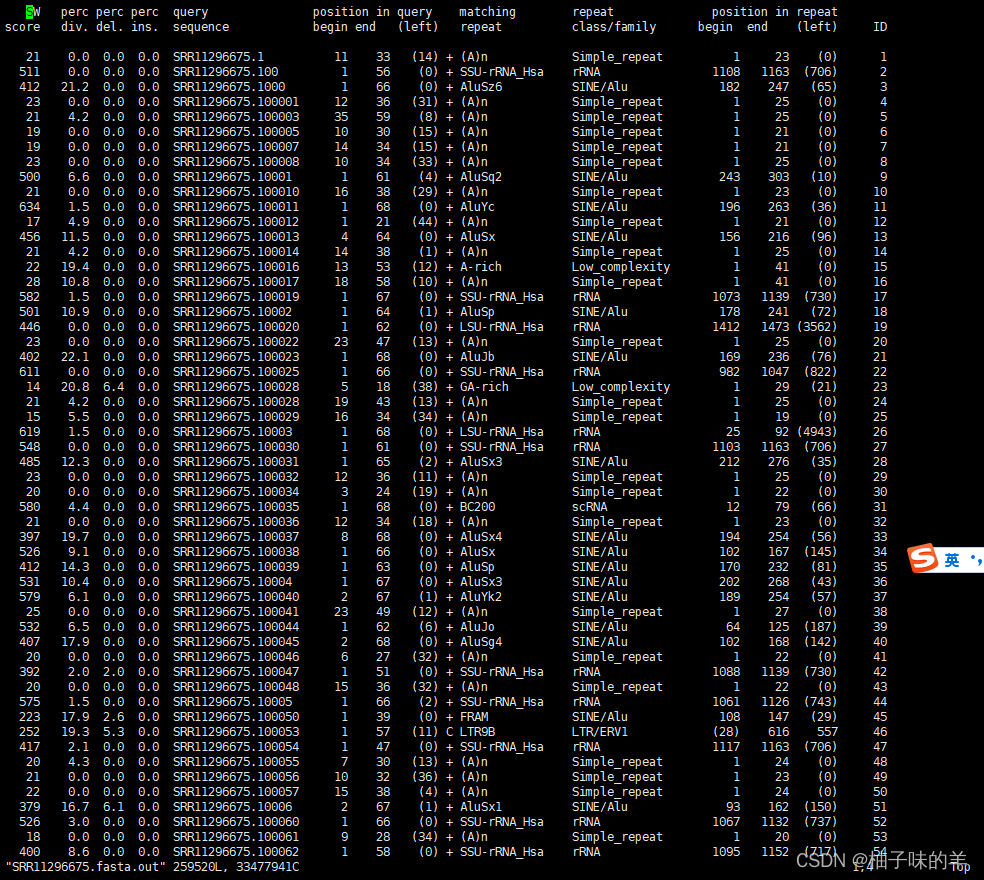
2. out文件
输出具体的repeat发生在基因上的位置,repeat的类型等
3. out.gff文件(和.out文件完全一样)
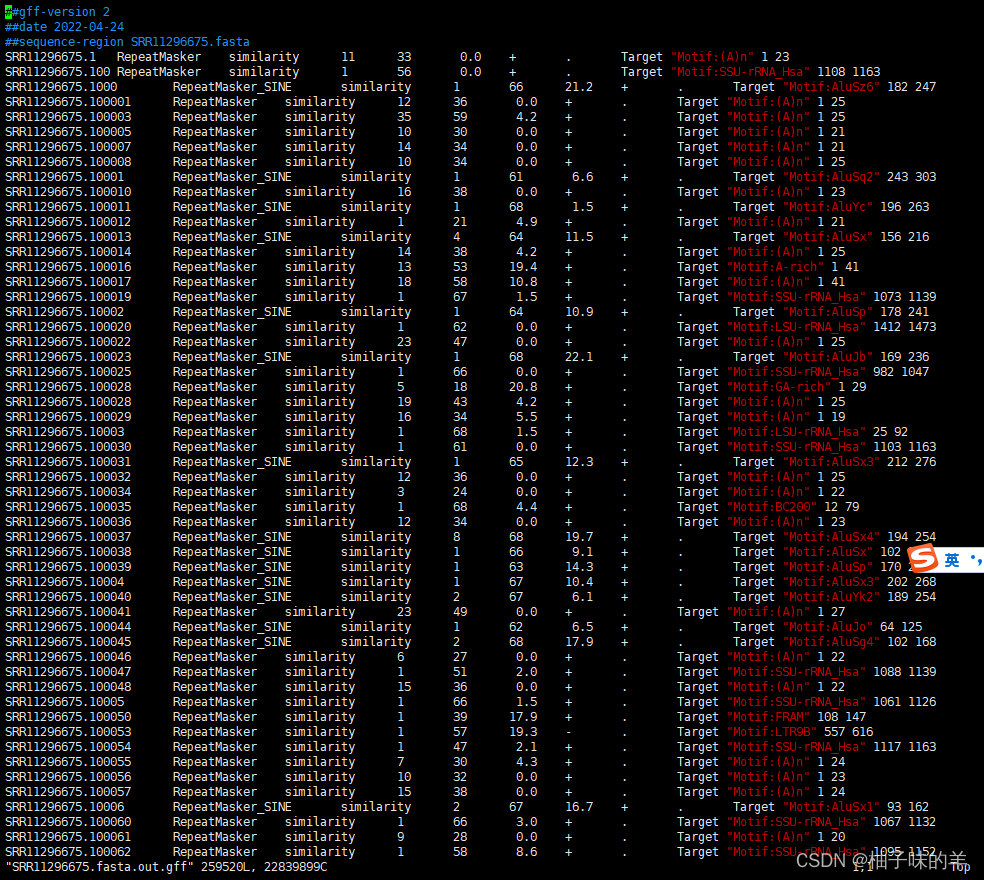
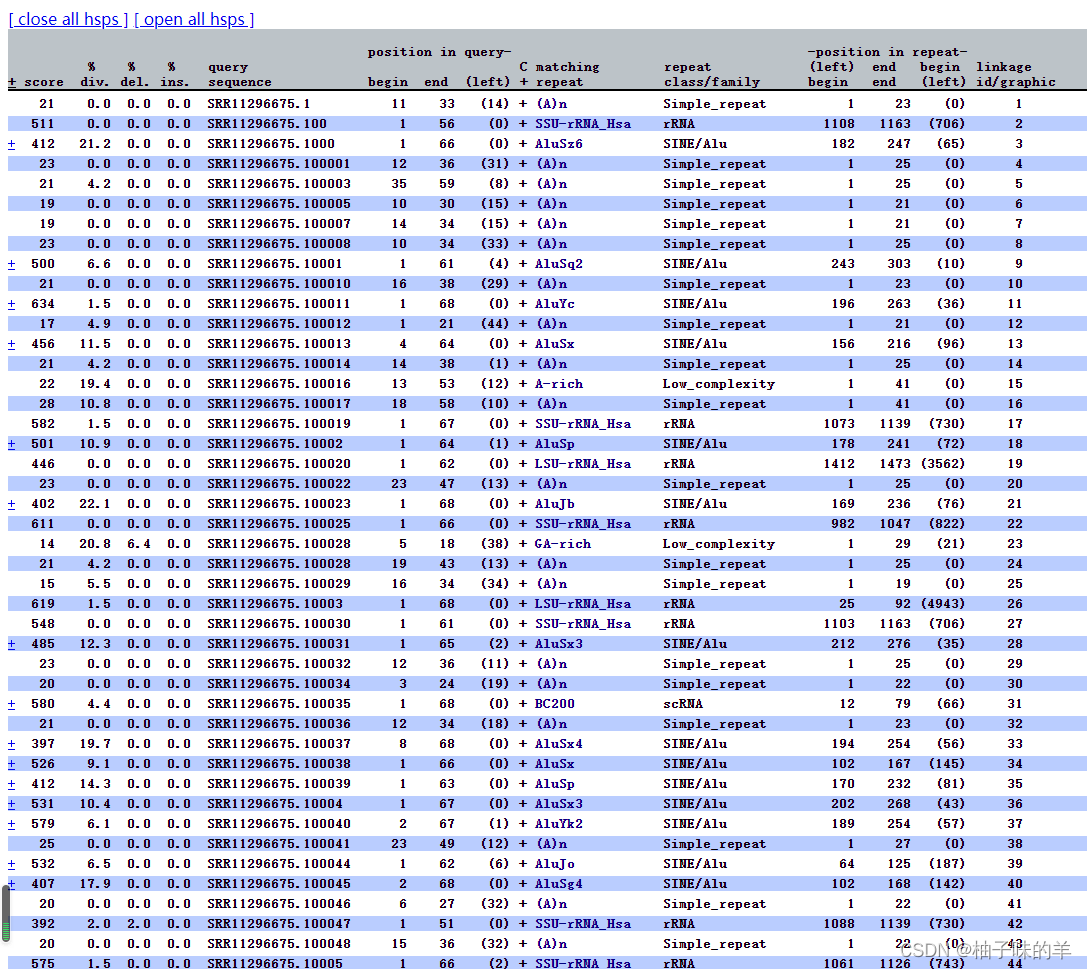
4. out.html文件(和.out文件一样,只是在html,显示方式不同罢了)
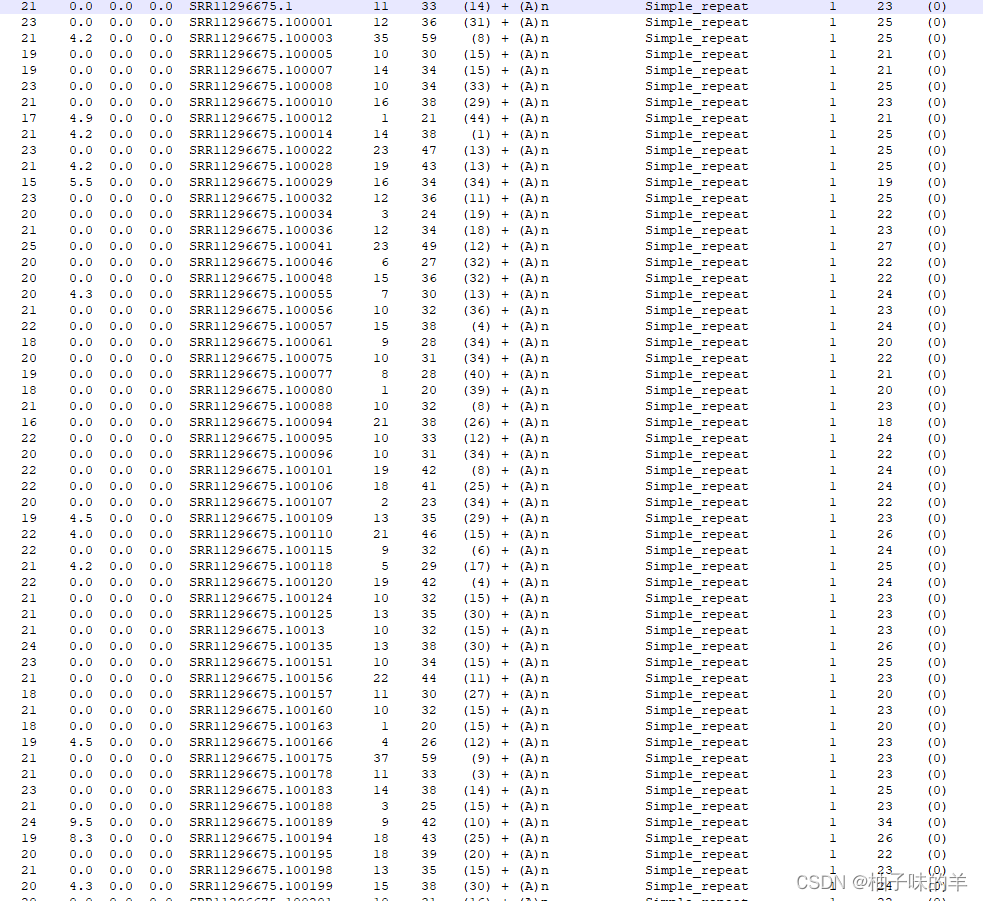
5. polyout文件——单独列出了微卫星表格
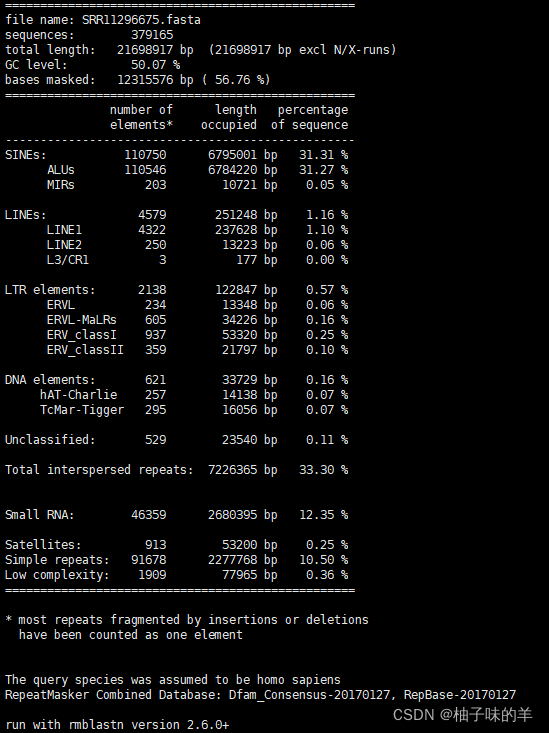
6. tbl文件——统计文件
其中的几个文件我不需要,比如out文件,out.gff文件和out.html文件都是一个东西,只是呈现方式不同,所以在跑正式数据的时候没有加-gff和-html

为开发者提供学习成长、分享交流、生态实践、资源工具等服务,帮助开发者快速成长。
更多推荐


 9
9 0
0- 0



 已为社区贡献3条内容
已为社区贡献3条内容

所有评论(0)