基因组组装---基因组大小评估(genome survey)
基因组组装---基因组大小评估(genome survey)
基因组组装---基因组大小评估(genome survey)
1. 估算基因组大小原理
kmer(k)是一段固定长度的短序列,因为对于长度为L的序列(L>k),每次移动一个字符,共有L - k +1个kmer序列。
此外,发现kmer在基因组大小评估和组装中为奇数,这是因为基因组的序列中存在回文序列(palindromic sequence),若为偶数的情况, 如,CGCGCGCG, kmer=4,这样他的互补链仍然是他的自身,这样产生的 de Bruijn graph很困难,不能区分这个序列是来自哪。总不能一会将他的正义链贴到基因组上,一会又将他的反义链再次贴到相同的位置上,也会造成组装困难。
举例:
L:GATCCTACTGATGC ( L = 14 )
kmer:8
L序列中kmer的总个数:
N = ( L - k ) + 1
= (14 - 8 ) + 1
= 7
GATCCTACTGATGC ( L = 14 )
GATCCTAC,
ATCCTACT,
TCCTACTG,
CCTACTGA,
CTACTGAT,
TACTGATG,
ACTGATGC
对于下面表格中的基因组而言,可以看到基因组只是覆盖了一次(整体覆盖,N=L - k + 1),对于1Mb基因组,估算误差已经很小了;假定测了多次(10 ×),那么就要计算总的kmer数然后除以覆盖度(10 ×)。
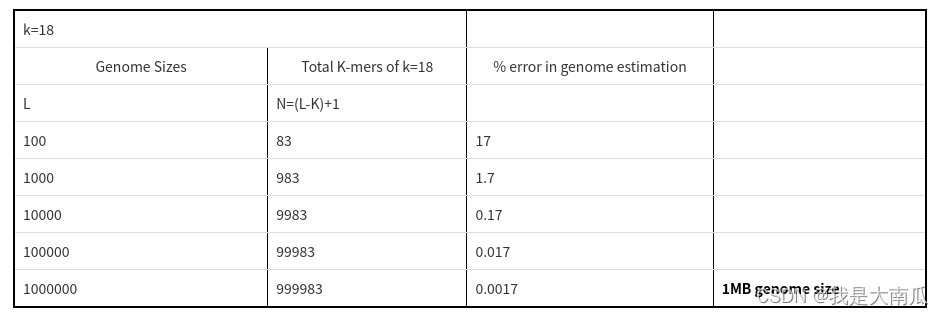
那么由此可推,对于更大的基因组序列,kmer的数值是设定的固定值,每个覆盖度(Coverage)对应着不同的数量(Number),这时,Total_kmer_number = Coverage (column) * Number (column),找到覆盖度的最大值(Expeacted_coverage,不算起初的error),也就是期望的平均覆盖度:
Genome_size = Total_kmer_number / Expected_coverage
2. Jellyfish软件
Genome survey的软件比较常用的有Jellyfish,其中子命令可以参看官网(https://github.com/gmarcais/Jellyfish/blob/master/doc/Readme.md)。
使用也很简单:
(1)统计kmer的命令是:
size=200M
kmer=17
fq1=/home/debian/data/08.arabidopsis_t2t_genome/CRR302670/CRR302670_r1.fastq.gz
fq2=/home/debian/data/08.arabidopsis_t2t_genome/CRR302670/CRR302670_r2.fastq.gz
jellyfish count -s ${size} -t 10 -m ${kmer} \
-C -o jellyfish_kmer${kmer}.count.txt\
--min-quality=20\
--quality-start=33\
<(zcat ${fq1})\
<(zcat ${fq2})
(2)根据(1)中统计结果计算kmer的histogram
jellyfish histo -t 10 jellyfish_kmer${kmer}.count.txt -o jellyfish_kmer$kmer.hist.txt
得到的结果可以使用genomescope软件汇总和绘图。
3. GenomeScope计算杂合度
GenomeScope软件可以将jellyfish结果拟合计算基因组杂合度信息:
(genomescope web网页http://qb.cshl.edu/genomescope/)
## 150 是PE read length
## ./genomescope_${kmer}_out 是输出文件夹
kmer=17
Rscript /home/debian/bin/genomescope-1.0.0/genomescope.R jellyfish_kmer${kmer}.hist.txt \
${kmer} 150 ./genomescope_${kmer}_out
4. GCE软件
GCE软件也是可以达到jellyfish的功能。
## 统计kmer
echo `date`;~/bin/GCE-master/gce-1.0.2/kmerfreq -k 17 -t 10 -p gce_kmer17 read.list; echo `date`
## 输出kmer统计
cat gce_kmer17.kmer.freq.stat| grep "#Kmer indivdual number"
cat gce_kmer17.kmer.freq.stat | perl -ne 'next if(/^#/ || /^\s/); print; ' | awk '{print $1"\t"$2}' >
kmer17.freq.stat.2colum
## 输出拟合估算基因组结果
~/bin/GCE-master/gce-1.0.2/gce -g 12235478628 -f kmer17.freq.stat.2colum >gce.table 2>gce.log
5. 估算拟南芥基因组大小
使用的二代测序数据是Illumina Novaseq 6000 PE 150(数据来源BIG GSA: CRA004538)
CRR302670_r1.fastq.gz ## 3.0 Gb
CRR302670_r2.fastq.gz ## 3.3 Gb
(1)使用 jellyfish + genomescope
设置jellyfish参数:
kmer=17 #($1),
size=200M #($2)
## genomescope目前只适用于二倍体基因组数据
整体代码为:
#!/usr/bin/env bash
# date: 2022.8.28 (22:55:03)
####################################################
############### INSTALL SOFTWARE ###################
####################################################
## Install new R (original R had png error...)
# mamba install -c conda-forge/label/cf202003 r-base
## Install genomescope
# wget -c https://github.com/schatzlab/genomescope/archive/refs/tags/v1.0.0.zip
# genomescope-1.0.0.zip
# unzip genomescope-1.0.0.zip
## Jellyfish count
## Install jellyfish
# mamba install -c bioconda kmer-jellyfish
# jellyfish 2.3.0
####################################################
########### Run jellyfish and genomescope ##########
####################################################
## set Illumina DNA files
kmer=$1 ## kmer number
size=$2 ## hash size (100M, 1G)
fq1=/home/debian/data/08.arabidopsis_t2t_genome/CRR302670/CRR302670_r1.fastq.gz
fq2=/home/debian/data/08.arabidopsis_t2t_genome/CRR302670/CRR302670_r2.fastq.gz
## run main process
echo `date` "count start"
## note `<()` had no space
jellyfish count -s ${size} -t 10 -m ${kmer} \
-C -o jellyfish_kmer${kmer}.count.txt\
--min-quality=20\
--quality-start=33\
<(zcat ${fq1})\
<(zcat ${fq2})
echo `date` "count end"
## jellyfish hist
echo `date` "histo start"
jellyfish histo -t 10 jellyfish_kmer${kmer}.count.txt -o jellyfish_kmer$kmer.hist.txt
echo `date` "histo end"
## GenomeScope estimate heterozygous rate
echo `date` "Genomescope start"
Rscript /home/debian/bin/genomescope-1.0.0/genomescope.R jellyfish_kmer${kmer}.hist.txt \
${kmer} 150 ./genomescope_${kmer}_out
echo `date` "Genomescope end"
查看评估结果信息:
cat ./genomescope_17_out/summary.txt
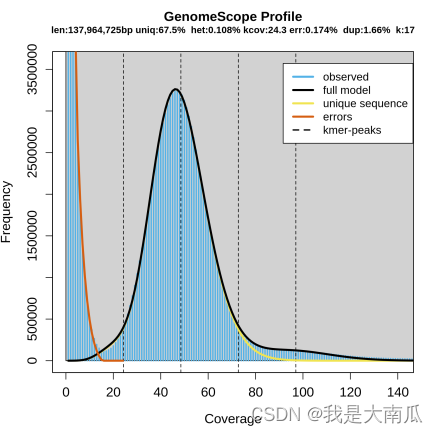
GenomeScope version 1.0
k = 17
property min max
Heterozygosity 0.106724% 0.109489%
Genome Haploid Length 137,917,082 bp 137,964,725 bp
Genome Repeat Length 44,864,650 bp 44,880,148 bp
Genome Unique Length 93,052,432 bp 93,084,577 bp
Model Fit 95.205% 97.5522%
Read Error Rate 0.174374% 0.174374%
genomescope统计杂合度结果和绘图:
绘图结果为plot.png和plot.log.png,前者为下图展示结果。
从下图可以看出明显的单峰分布,说明杂合度很低,仅有0.108%,估算基因组大小为 137,964,725 bp,基本复合预期。
(2)使用 GCE
GCE软件的整体命令在上面有提到,分开看kmer统计结果,其中12235478628需要用到gce主命令中:
head gce_kmer17.kmer.freq.stat
#Kmer size: 17
#Maximum Kmer frequency: 65535
#Kmer indivdual number: 12235478628
#Kmer species number: 604346515
#Theoretic space of Kmer species: 17179869184 occupied ratio: 0.0351776
GCE软件的结果可以查看gce.log
Final estimation table:
raw_peak effective_kmer_species effective_kmer_individuals coverage_depth genome_size a[1] b[1]
62 101912174 11436641691 62.8413 1.81993e+08 0.906038 0.63522
Discrete mode estimation finished!
本GCE结果中估算的拟南芥基因组结果偏大 1.81993e+08 bp,为182Mb,可能是统计时候未进行过滤吧。
重新使用fastp软件过滤:
#!/usr/bin/bash
#### Run fastp version 0.22.0
kmerfreq=~/bin/GCE-master/gce-1.0.2/kmerfreq
gce=~/bin/GCE-master/gce-1.0.2/gce
echo `date` fastp start
fastp -i /home/debian/data/08.arabidopsis_t2t_genome/CRR302670/CRR302670_r1.fastq.gz\
-I /home/debian/data/08.arabidopsis_t2t_genome/CRR302670/CRR302670_r2.fastq.gz\
-o CRR302670.clean.R1.fq.gz -O CRR302670.clean.R2.fq.gz\
-q 20 -w 10 -h CRR302670.fastp_filter.html
echo `date` fastp end
#### Run GCE version 1.0.2
ls CRR302670.clean.R*.fq.gz > read_file
$kmerfreq -k 17 -t 20 -p mucao_k17 read_file
less mucao_k17.kmer.freq.stat | grep "#Kmer indivdual number"
less mucao_k17.kmer.freq.stat | perl -ne 'next if(/^#/ || /^\s/); print; ' | awk '{print $1"\t"$2}' > mucao.kmer.freq.stat.2colum
## Get kmer number
num=`head -n 3 mucao_k17.kmer.freq.stat | tail -n1 | cut -f 4 -d " "`
## Common first 10 lines contains much error results
depth=`sed '1,10d' kmer17.freq.stat.2colum |awk 'NR==1{max=$2;next}{max=max>$2?max:$2}END{print max}'`
$gce -g $num -f ./mucao.kmer.freq.stat.2colum -c $depth >gce.table 2>gce.log
估算基因组大小结果查看(gce.log),此时基因组大小为179 Mb,还是较大。
Final estimation table:
raw_peak effective_kmer_species effective_kmer_individuals coverage_depth genome_size a[1]b[1]
62 101924949 11259729986 62.7518 1.79433e+08 0.907537 0.626289
Discrete mode estimation finished!
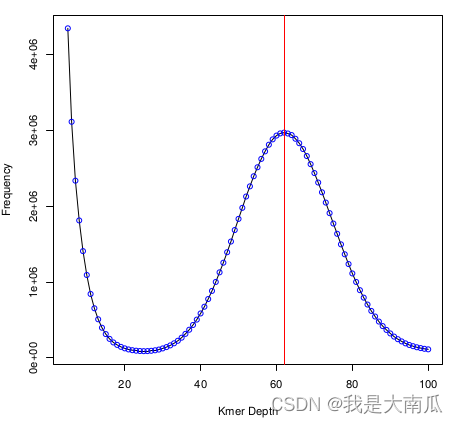
使用mucao.kmer.freq.stat.2colum数据,可以绘制kmer分布图(jellyfish的histo结果也可以绘制):
data=read.table("mucao.kmer.freq.stat.2colum",header=F)
a1=data[5:100,]
a2=data[20:100,]
peakNum=a2[a2$V2==max(a2$V2),][,1]
d=a1
pdf(file = "GCE_kmer_dis.pdf")
plot(d[,1],d[,2],type="l",xlab="Kmer Depth", ylab="Frequency")
points(d[,1],d[,2],col="blue")
abline(v=peakNum,col="red")
dev.off()
6. 总结
(1)就测试数据而言,jellyfish + genomescope使用体验更好,估算基因组大小更符合实际(ensemble plants arabidopsis ~ 135Mb);本数据GCE结果偏大, GCE中参数还特别设定纯和和杂和模式,更加人性化。
(2)kmer估算基因组大小,通过上面方法外,还可以使用初步组装结果表征,或者通过流式细胞仪实验估计。
参考:
https://bioinformatics.uconn.edu/genome-size-estimation-tutorial/ (kmer估算基因组大小原理)
https://bioinformatics.stackexchange.com/questions/156/why-do-some-assemblers-require-an-odd-length-kmer-for-the-construction-of-de-bru (kmer是奇数)
https://github.com/gmarcais/Jellyfish/blob/master/doc/Readme.md (Jellyfish软件)
https://github.com/schatzlab/genomescope (GenomeScope软件)
https://bioinformaticsworkbook.org/dataAnalysis/GenomeAssembly/genomescope.html#gsc.tab=0 (GenomeScope 软件说明)
https://github.com/fanagislab/GCE (GCE软件)

为开发者提供学习成长、分享交流、生态实践、资源工具等服务,帮助开发者快速成长。
更多推荐



 4
4 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)