R统计-PCA/PCoA/db-RDA/NMDS/CA/CCA/DCA等排序分析教程
,这个zhe 点击名片 关注我们一、定义oridination(gradient analysis):研究群落之间、群落与成员之间、群落与其环境之间的复杂关系。排序的过程是将样方或物种...
,这个zhe
点击名片 关注我们
一、定义
oridination(gradient analysis):研究群落之间、群落与成员之间、群落与其环境之间的复杂关系。排序的过程是将样方或物种排列在一定的空间,使得排序轴能够反映一定的生态梯度。在一个可视化的低维空间重新排列样方,使得样方之间的距离最大程度地反映出平面散点图内样方之间的关系信息。
二、分类
2.1 根据物种响应环境梯度模型分类
使用线性响应模型的排序方法叫线性排序(linear ordination),包括RDA和PCA等;而基于单峰响应模型的被称为非线性排序(nonlinear ordination),包括CCA、CA、DCA和DCCA等。线性响应模型通常使用最小二乘法进行回归拟合。单峰响应模型则是通过基于所有包含该物种的样方中环境因子的加权平均得到该物种在环境梯度上的最适値。单峰模型能更好的反映种-环境和种-种之间的关系。在具体分析的时候怎样选择模型,参考R绘图-RDA排序分析一文。需要补充的是物种数据的量纲不同时不适合做单峰模型排序;有空样方出现的数据要采用单峰分析,需要把空样方剔除。
2.2 根据是否使用环境因子数据分类
约束排序是在已有环境因子中寻找最相关的解释群落结构变化的变量,所用环境因子数要至少要比样品数少2;而非约束排序是寻找最能展示群落结构变化的潜在的解释变量,以排序轴的形式体现。
2.2.1 unconstrained ordination(非约束排序/间接排序) :只使用物种组成数据的排序
(1)principal components analysis:主成分分析(PCA),基于原始的物种组成矩阵所做的排序分析。
(2)correspondence analysis:对应分析(CA)
(3)Detrended correspondence analysis:去趋势对应分析(DCA)
(4)principal coordinate analysis:主坐标分析(PCoA) ,基于由物种组成计算得到的距离矩阵得出的。
(4)non-metric multi-dimensional scaling:非度量多维尺度分析(NMDS)
2.2.2 constrained ordination(约束排序/直接排序):使用物种和环境因子数据排序
(1)redundancy analysis:冗余分析(RDA)
(2)Canonical correspondence analysis:典范对应分析(CCA)
(3)Detrended canonical correspondence analysis:降趋势典范对应分析(DCCA)
(4)Canonical variate analysis:典型变量分析(CVA)
(5)distance based redundancy analysis (db-RDA):基于距离的冗余分析
2.3 偏分析(partial analysis)
预先移除群落结构变化中由协变量产生的那部分影响,在通过排序展示剩下的变化量的排序方法,约束排序和非约束排序均有相应的偏分析方法。
2.3.1 Partial methods of unconstrained ordination:非约束性偏分析方法
partial PCA;partial CA ;partial DCA
2.3.2 Partial methods of constrained ordination:约束性偏分析方法
partial RDA;partial CCA;partial DCCA;partial CVA
三、R中进行排序分析
Canoco和R中的vegan包都可以实现上述排序分析,但是Canoco是一个收费软件,而且不能像R中ggplot2绘制精美的图,因此下面我使用R来展示排序分析。
3.1 数据准备
所有排序分析都使用的是同一套数据,一个不同放牧处理的土壤微生物物种组成数据,对应样品的土壤理化因子数据以及一个分类因子文件。
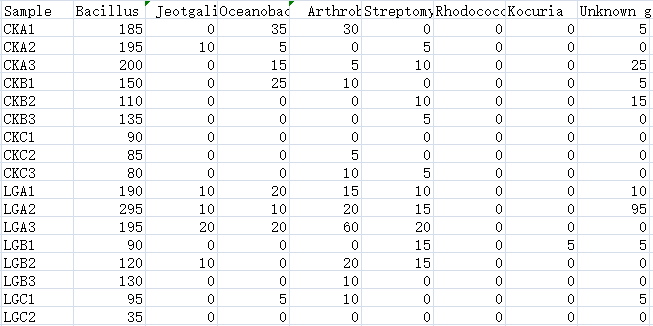
图1|物种组成数据。

图2|环境因子数据。

图3|分类数据。
# 1. 设置工作路径,老生常谈,设置工作路径和查看路径内容一定是第一步
setwd("D:\\ordination")
getwd()
dir()
# 2. 调用R包
library(vegan)
library(ecodist)
library(ggplot2)
# 3. 读入数据
spe=read.csv("spe.csv",header=TRUE,row.names=1,sep=",", colClasses = c("character",rep("numeric",times=8)) )# 群落组成数据,物种组成数据是整数,为了使读入数据格式为数值型,设置colClasses,7为物种种类。
## ?rep # 查看函数用法和函数中参数意义
spe
group=read.csv("group.csv",header=TRUE,row.names=1,sep=",")#分类数据,包含两种类别,grazing和soil depth
group
env=read.csv("env.csv",header=TRUE,row.names=1,sep=",") #环境数据:土壤理化因子
env
3.2 PCA
# PCA和RDA分析都使用的是vegan::rda()函数,不加环境因子就是PCA分析
## 基于线性模型的排序方法会涉及数据的中心化与标准化,
## 中心化:每个数据减去平均值,让每个样品/物种的均值为0;
## 标准化:让每个数据除以均方根,让每个样品/物种的方差为1.
## 中心化与标准化不是必须进行的,几乎基于线性模型的数据都需要中心化,但不一定需要标准化,中心化必须在标准化前面进行。
## 如果物种数据的单位不同,则必须进行标准化
pca=rda(spe,scale=T) # scale=T,表示单位方差标准化,先中心化再均值化,物种数据均值为0,方差为1.
pca
## 绘图数据提取
s.pca=pca$CA$u # 提取样本特征值
s.pca
e.pca=pca$CA$v # 提取物种特征值
e.pca # 可执行选取排序轴绘制散点图
eig=pca$CA$eig # 提取特征根,计算PC1和PC2的解释度,横纵坐标标签

图4|PCA的描述统计结果。

图5|样本特征值。每个排序轴所负荷的特征值,可以认为是每个排序轴所能解释的方差变化的量。

图6|物种特征值。
3.3 RDA
RDA详细分析及绘图过程见R绘图-RDA排序分析一文。
# vegan::rda()函数,加环境因子就是RDA分析
RDA=rda(spe,env,scale=TRUE)
RDA
summary(RDA)
## 绘图数据提取
s.RDA=RDA$CCA$u # 提取样本特征值
s.RDA
e.RDA=RDA$CCA$v # 提取物种特征值
e.RDA # 可执行选取排序轴绘制散点图
env.RDA=RDA$CCA$biplot # 提取环境因子特征值
env.RDA
# 检查环境因子相关性
## 整个环境因子对群落变化相关性的显著性
RDA.perm=permutest(RDA,permu=999)
RDA.perm
## 检查每个环境因子与群落变化的相关性
RDA.env=envfit(RDA,env,permu=999)
RDA.env
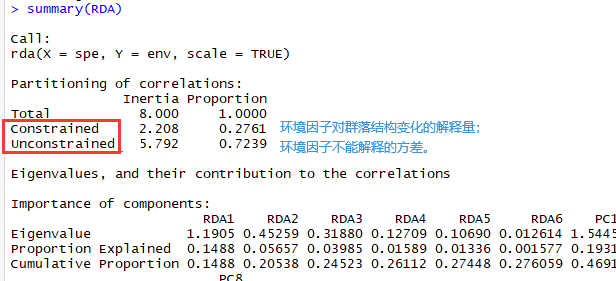
图7|RDA描述统计结果。

图8|环境因子特征值。

图9|环境因子与群落结构变化的相关性。
3.4 CA
# CA与CCA都是使用vegan::cca()函数,不加环境因子就是CA分析
CA=cca(spe,env,scale=TRUE)
CA
summary(CA)
## 绘图数据提取
## 绘图数据提取
s.CA=CA$CA$u # 提取样本特征值
s.CA
e.CA=CA$CA$v # 提取物种特征值
e.CA # 可执行选取排序轴绘制散点图

图10|CA描述统计结果。
3.5 CCA
# vegan::cca()函数,加环境因子就是CCA分析
CCA=cca(spe,env)
CCA
summary(CCA)
## 绘图数据提取
s.CCA=CCA$CCA$u # 提取样本特征值
s.CCA
e.CCA=CCA$CCA$v # 提取物种特征值
e.CCA # 可执行选取排序轴绘制散点图
env.CCA=CCA$CCA$biplot # 提取环境因子特征值
env.CCA
# 检查环境因子相关性
## 整个环境因子对群落变化相关性的显著性
CCA.perm=permutest(CCA,permu=999)
CCA.perm
## 检查每个环境因子与群落变化的相关性
CCA.env=envfit(CCA,env,permu=999)
CCA.env

图11|CCA描述解读结果。
3.6 PCoA
PCoA(主坐标分析)跟PCA不同的是,它需要先计算一个距离矩阵,经过投影后,在低维度空间进行距离展示,以最大限度地保留原始样本的距离关系,使相似的样本在图形中的距离更为接近,相异的样本距离更远。PCA则使用的是原始的样品X物种矩阵,是设法保留数据里的变异,让点的位置尽量不要变动。
# 使用vegdist()函数求样品间距离矩阵
spe.dist.bray<-vegdist(spe,method = 'bray') # 距离矩阵计算有多种方法可以选择,"manhattan", "euclidean", "canberra", "clark", "bray", "kulczynski", "jaccard", "gower", "altGower", "morisita", "horn", "mountford", "raup", "binomial", "chao", "cao", "mahalanobis", "chisq" or "chord".默认是bray
spe.dist.bray
# 将距离矩阵进行主坐标分析
pcoa = cmdscale(spe.dist.bray, k=3, eig=TRUE) # k设置坐标轴数量,推荐用3或者样本数-1; eig=TRUE返回特征根,默认不输出特征根结果
pcoa
# 提取样品坐标信息
points=scores(pcoa) # 可以提取的同时形成数据框
## points = as.data.frame(pcoa$points) # 也可以提取坐标信息
points = as.data.frame(pcoa$points)
# wascores()函数以多度加权平均方式将物种被动投影到样方的PCoA图中,获得物种坐标信息
spe.wa <- wascores(pcoa$points, spe)
spe.wa
# 提取特征根
eig = pcoa$eig
eig
# 绘制PCoA排序图,其实排序分析做完之后,都是使用ggplot2根据坐标信息绘制散点图就行,这里以PCoA为例子画一个图吧。
points = cbind(points, group[match(rownames(points), rownames(group)), ])
points
cols=c("CK"="grey","LG"="red","MG"="blue","HG"="green")
treats=as.vector(points[,4])
treats
depth=as.vector(points[,5])
depth
points=data.frame(points)
points
p = ggplot(points, aes(x=Dim1, y=Dim2, colour=treats,shape=depth)) +scale_colour_manual(values=cols,breaks=c("CK","LG","MG","HG"))+theme(legend.title=element_blank())+scale_fill_manual(values=cols,breaks=c("CK","LG","MG","HG"))+
geom_point(alpha=.7, size=4) + scale_shape_manual(values=c(15,16,17))+geom_hline(yintercept=0) + geom_vline(xintercept=0)+theme_bw() + theme(panel.grid=element_blank()) +
labs(x=paste("PCoA 1 (", format(100 * eig[1] / sum(eig), digits=4), "%)", sep=""),
y=paste("PCoA 2 (", format(100 * eig[2] / sum(eig), digits=4), "%)", sep=""),
title="bray_curtis PCoA")
p

图12|bray-curtis距离矩阵。

图13|物种投影数据。

图14|points样品坐标与分类数据合并表.

图15|PCoA排序图。
3.7 NMDS
非度量多维尺度法是一种将多维空间的研究对象(样本或变量)简化到低维空间进行定位、分析和归类,同时又保留对象间原始关系的数据分析方法。适用于无法获得研究对象间精确的相似性或相异性数据,仅能得到他们之间等级关系数据的情形。其基本特征是将对象间的相似性或相异性数据看成点间距离的单调函数,在保持原始数据次序(秩)关系的基础上,用新的相同次序的数据列替换原始数据进行度量型多维尺度分析。换句话说,当资料不适合直接进行变量型多维尺度分析时,对其进行变量变换,再采用变量型多维尺度分析,对原始资料而言,就称之为非度量型多维尺度分析。其特点是根据样品中包含的物种信息,以点的形式反映在多维空间上,而对不同样品间的差异程度,则是通过点与点间的距离体现的,最终获得样品的空间定位点图。
# NMDS
#distance.bray<-vegdist(spe,method = 'bray')
#NMDS<-metaMDS(distance.bray,k=3)
NMDS<-metaMDS(spe,distance = "bray",k=3) # k设置维度数
NMDS
# 提取样本坐标数据
NMDS$points
# NMDS胁迫系数stress:检验NMDS分析结果的优劣,stress<0.2,可用NMDS的二维点图表示,其图形有一定解释意义;stress<0.1,认为是一个好的排序;stress<0.05,具有很好的代表性。
NMDS$stress

图16|NMDS结果描述统计。
3.8 DCA
# DCA,可用于判别使用线性模型还是单峰模型
DCA=decorana(spe)
DCA
# 查看样品和物种得分
all.DCA=summary(DCA)
all.DCA$spec.scores # 物种得分
all.DCA$site.totals #样本得分
s.DCA=scores(DCA) # 仅看样品得分
DCA$evals # 查看轴特征根

图17|DCA描述统计结果。
3.9 db-RDA
# db-RDA,vegan包中有直接运行的函数
dbRDA=dbrda(spe ~ ., env, dist="bray")
## 也可以挑选部分环境因子进行dbRDA分析
# partdbRDA=dbrda(spe ~ pH+WC, env, dist="bray")
dbRDA
# 后续提取样本、物种分数、特征值以及进行环境因子与群落变化相关性的方法与RDA一样,这里不在赘述
# 使用anova()进显著性检验
test=anova(dbRDA, by="terms", permu=999)
test
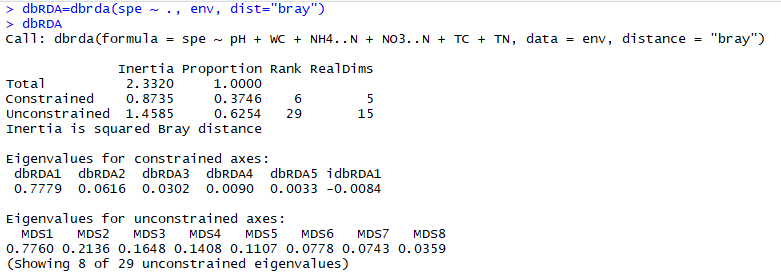
图18|db-rda结果描述统计

图19|ANOVA检验结果
4.0 偏分析
偏分析方法可以分别分析主环境变量与协变量对群落结构的影响。下面以RDA为例进行偏分析。
# partial-RDA
pRDA=rda(spe,env[1:4],env[5:6]) # env[1:4]主环境变量矩阵(Constraining matrix),env[5:6]协变量矩阵(Conditioning matrix).
pRDA
# 提取分析结果方法同RDA相同,这里就不在赘述了。
summary(pRDA)
pRDA$pCCA # 协变量结果
pRDA$CCA # 主环境变量结果
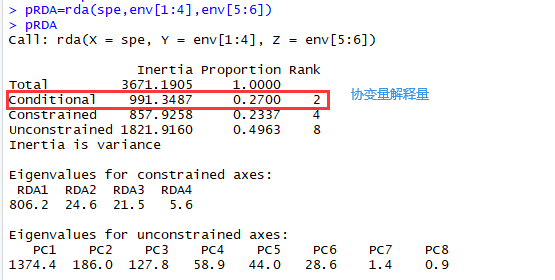
图20|pRDA分析结果。
需要原始数据和R脚本的同学或老师,后台留言“R统计-排序分析”即可获得。
参考文献
排序方法差异:http://blog.sciencenet.cn/home.php?mod=space&uid=769877&do=blog&id=1080370
三文读懂PCA与PCoA:https://www.genewiz.com.cn/zh-CN/Public/Resources/zxzx/0009
添加物种投影:https://www.jianshu.com/p/12a5abbef81f
关于冗余分析(RDA)中环境因子共同解释部分出现负值的说明:http://blog.sciencenet.cn/blog-267448-1187530.html
关于RDA中每个环境因子解释率的说明:http://blog.sciencenet.cn/blog-267448-1186999.html
ggplot2颜色:http://www.cookbook-r.com/Graphs/Colors_(ggplot2)/
推荐阅读
开放转载
公众号原创文章开放转载,在文末留言告知即可
EcoEvoPhylo :主要分享微生物生态和phylogenomics的数据分析教程。点击上方名片,即可关注EcoEvoPhylo。让我们大家一起学习,互相交流,共同进步。

为开发者提供学习成长、分享交流、生态实践、资源工具等服务,帮助开发者快速成长。
更多推荐



 16
16 0
0- 0



 已为社区贡献7条内容
已为社区贡献7条内容

所有评论(0)