Nature综述:宏基因组测序研究耐药基因的方法和资源
本文转自红皇后学术,链接https://mp.weixin.qq.com/s/2QMrq6hwr4mIPSpe_rfXJg论文信息论文题目:Sequencing-based method...

本文转自红皇后学术,链接 https://mp.weixin.qq.com/s/2QMrq6hwr4mIPSpe_rfXJg
论文信息
论文题目:Sequencing-based methods and resources to study antimicrobial resistance
期刊:Nature Reviews Genetics
IF:35.898
发表时间:2019
链接:https://www.nature.com/articles/s41576-019-0108-4
摘要
抗菌素耐药性使得细菌对抗生素产生免疫力,这使得疾病的发病率和死亡率以及治疗的成本大量增加。
识别和理解抗菌素耐药性对于治疗耐药菌感染以及限制耐药菌扩散的临床和公共卫生实践至关重要。
新一代测序技术正在扩展我们检测和研究抗菌素耐药性的能力,本篇综述包含从传统的抗生素敏感性测试到最新的深度学习方法,详细介绍了抗菌素耐药性的鉴定和表征方法。
本篇文章专注于基于测序的耐药性发现,并讨论了用于抗菌素耐药性研究的工具和数据库。
引言
抗菌素是可以抑制或杀死细菌的小分子,这些小分子通常被用于治疗细菌感染,但是尽管有抗菌素的压力,些细菌仍能生长和存活,这种特性被称为抗菌素抗性。
在临床环境中,与易感细菌引起的感染相比,耐药菌感染会降低可用的治疗方法,同时增加发病率和死亡率。
目前,几乎所有的抗菌素都已经发现了细菌对其产生的抗药性,甚至包括那些用于回威胁生命的、对多种药物具有抗药性的细菌感染中使用的所谓的“最后手段”的抗菌药物。
在美国,每年有200万人会面临对临床一线使用的抗菌素具有耐药性的细菌感染,这些感染要花费200亿美元的医疗费用。这个问题并非只存在于美国,在欧盟,抗菌素耐药性导致超过30,000人死亡,近90万的疾病调整生命年。
实际上,多个国家和全球公共卫生组织抗菌素耐药性归为迫在眉睫的危险,并一致认为,追踪其耐药性和流行对最大限度地减少其对人类健康的威胁至关重要。
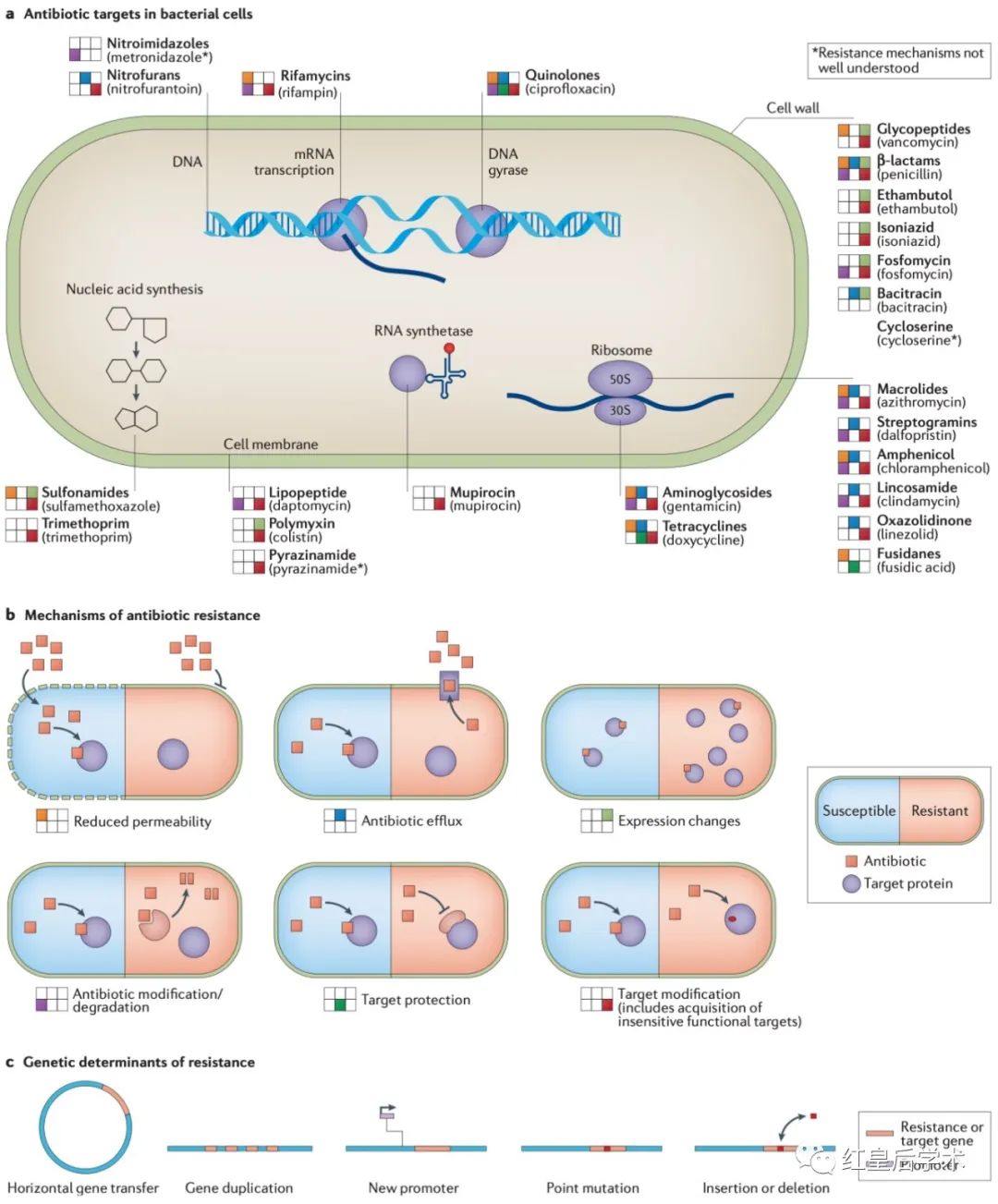
抗菌素药敏试验(AST)是测定细菌中抗菌素耐药性的传统方法,这些基于培养的测试确定了在存在抗菌素的情况下细菌的生长状况,AST被广泛用于医院临床微生物学实验室,因为它提供了可操作的表型耐药性数据以指导患者的治疗决策。
尽管基于培养的耐药性测定可以为患者管理和耐药性基因流行病学的研究提供重要的信息,但它在实施和信息内容上存在缺陷,进行AST需要微生物学设施和经过培训的临床微生物学人员以确保准确性,此外,AST仅对可培养细菌有效,这使得该方法无法研究那些不可培养的细菌占很大比例的各种复杂微生物群落中耐药性的出现和传播。
细菌的耐药性通常是由编码基因确定的,抗菌素药耐药性可通过多种机制获得,包括现有基因的过表达或复制、点突变以及通过水平基因转移(HGT)获得全新基因。
新一代测序技术和计算方法的进展促进了基因组和宏基因组中抗菌素耐药基因的快速鉴定和表征,这些发展中的技术和方法补充了用于临床和监测应用的传统的基于培养的方法,并为快速、准确地确定可培养和不可培养细菌的耐药性提供了机会。
对人类、动物和环境样品的大规模比较研究为抗菌素耐药性基因的全球分布、多药物抗性细菌的传播、耐药性交换网络以及以及不同的行为和系统发育如何影响全球抗菌素耐药性的演变提供了新的见解。
使用测序数据了解和调查耐药性的遗传决定因素是一种全新的挑战,我们可以通过改进计算算法以组织基因组数据并预测抗菌素耐药性或者通过发展非原位测序的技术来解决这些问题。
在本篇综述中,作者讨论了研究耐药性的各种方法的优缺点,以及用于基因组和宏基因组样品中耐药性基因鉴定的计算策略和资源,作者还描述了解决耐药性检测方法中缺点的最新进展,并介绍了需要重点关注的研究领域。
Box1:基于培养的药敏测试
细菌培养作为临床微生物学的组成部分具有悠久的历史,研究人员在琼脂平板或液体肉汤中培养细菌,以探测细菌表型并发现新的细菌功能,医院临床微生物学实验室可以使用从这些测定中获得的数据来指导临床治疗决策。
目前的技术
对于表型测试,通过非选择性或选择性琼脂平板从患者或环境样品中分离细菌,然后,将分离的细菌直接使用抗菌素刺激,以测试其耐药性。
在基于液体培养基的AST中,会通过不断降低抗菌素的使用浓度以找到细菌能够生长的最大浓度,抑制细菌生长的最低抗菌素浓度称为最低抑制浓度。
固体培养基技术使用Kirby-Bauer圆盘扩散法或梯度扩散条来测量细菌生长距离抗菌源的程度,抗菌圆盘或条带周围的区域称为抑制区。
临床和实验室标准协会(CLSI)或欧洲AST委员会(EUCAST)会基于测定的最小抑菌浓度或抑菌圈区域发布的耐药性确定的标准。
目前,不依赖测序的耐药性确定方法同样也有新的进展,其中包括基质辅助激光解吸/电离飞行时间质谱(MALDI–TOF-MS)、荧光原位杂交(FISH)和基于微流控的技术,这些技术可将AST的测试时间降低至30分钟,这些快速的测试技术对于生长缓慢的微生物特别有价值,例如结核分枝杆菌,传统的AST方法可能需要几个星期才能得到测试结果。
挑战和不足
尽管AST可用于提供表型耐药性结果,但它的通量很低,对于每个样品,临床微生物学家都需要培养细菌并建立和读取AST结果。
这存在一定的局限性,因为并非所有的医疗保健中心都对人员进行了临床微生物学相关的培训,此外对于某些细菌(例如结核分枝杆菌),目前在一些资源贫乏地区的实验室诊断技术灵敏度依然较低。
同时,目前发布的药敏标准还不能完全覆盖所有抗菌素和引起感染的可培养细菌的所有组合,不同国家之间的标准也不统一。
此外,在多种细菌可能导致疾病感染的情况下,基于培养的技术可能无法得到准确的结果,来自患者样本的一些宏基因组学研究表明,基于培养技术检测到的细菌可能与疾病症状无关。
最后,与全基因组测序相比,这些表型方法在检测耐药性决定因素方面具有较低的分辨率,并且有关耐药性基因流行病学的信息相对较少。
创新
自动化技术的快速发展和应用产生了多个基于表型分析的高效分析系统,包括VITEK、ADAGIO、Pheno在内的几种系统已经进入临床领域。
自动化平台具有几个关键优势,包括连续监控系统、用于测量药敏结果的灵敏光学仪器和标准化的内部时钟,这些优势可以加快获取培养细菌的药敏结果。它们还可以提高不同地区药敏结果的一致性,并减轻临床微生物学家的工作负担,不幸的是,这些自动化平台的建立成本过高。
基于测序的耐药性识别
测序技术的发展提高了细菌序列数据的可用性,并且不断降低的成本使测序成为了一种可行的抗菌素耐药性监测工具,近年来,已经发布了几种方法和工具,用于从全基因组测序和宏基因组测序数据中检测抗菌素耐药性的遗传决定因素。
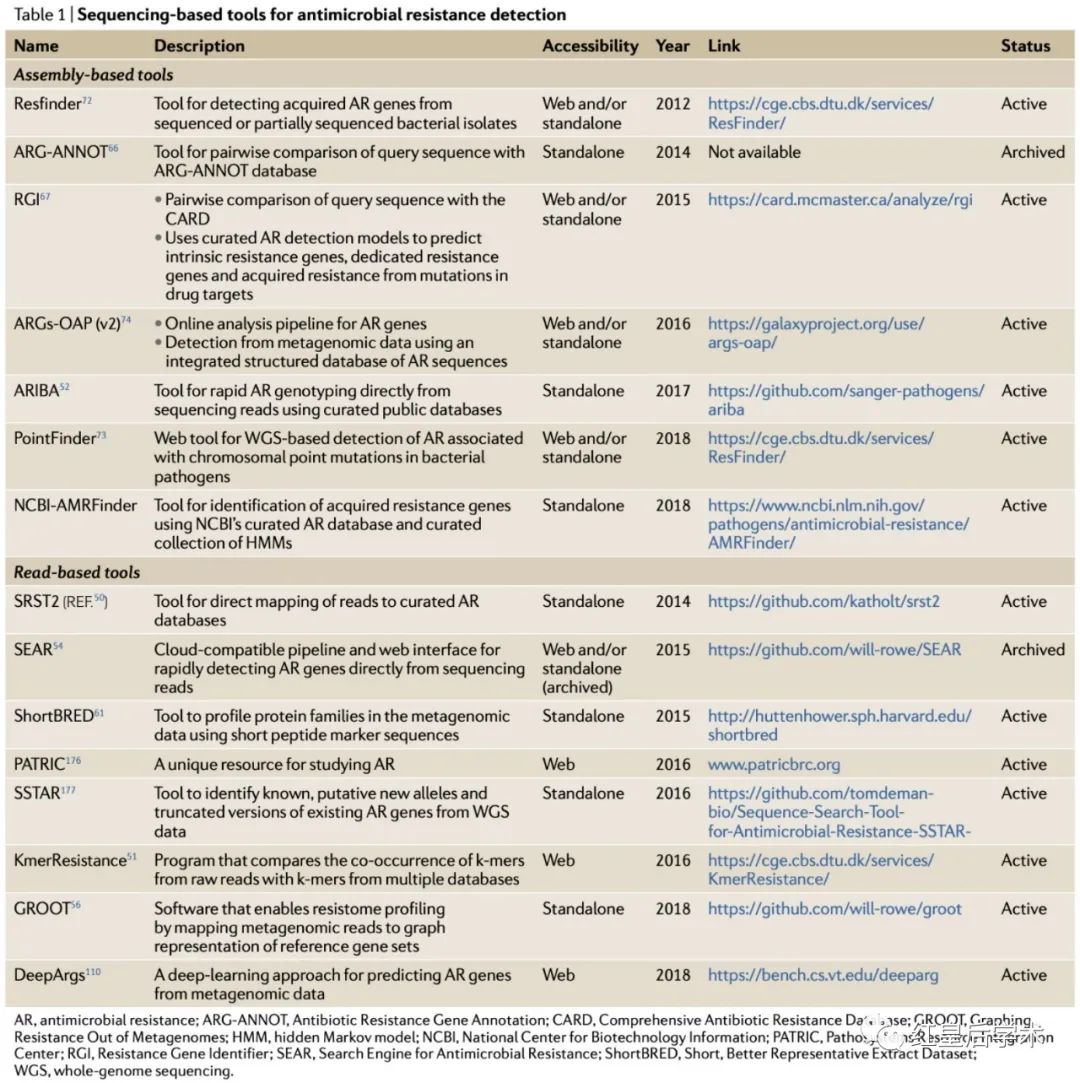
有效组织测序数据是抗菌素耐药性基因分析之前的重要预处理步骤,由Illumina等测序技术产生的短读段可以使用基于拼接的方法进行处理,从而先将测序读段组装成连续的片段(contig),然后通过与自定义或公共参考数据库进行比较进行注释,或者使用基于read的方法,通过将读数直接映射至参考数据库来预测耐药性决定因素。
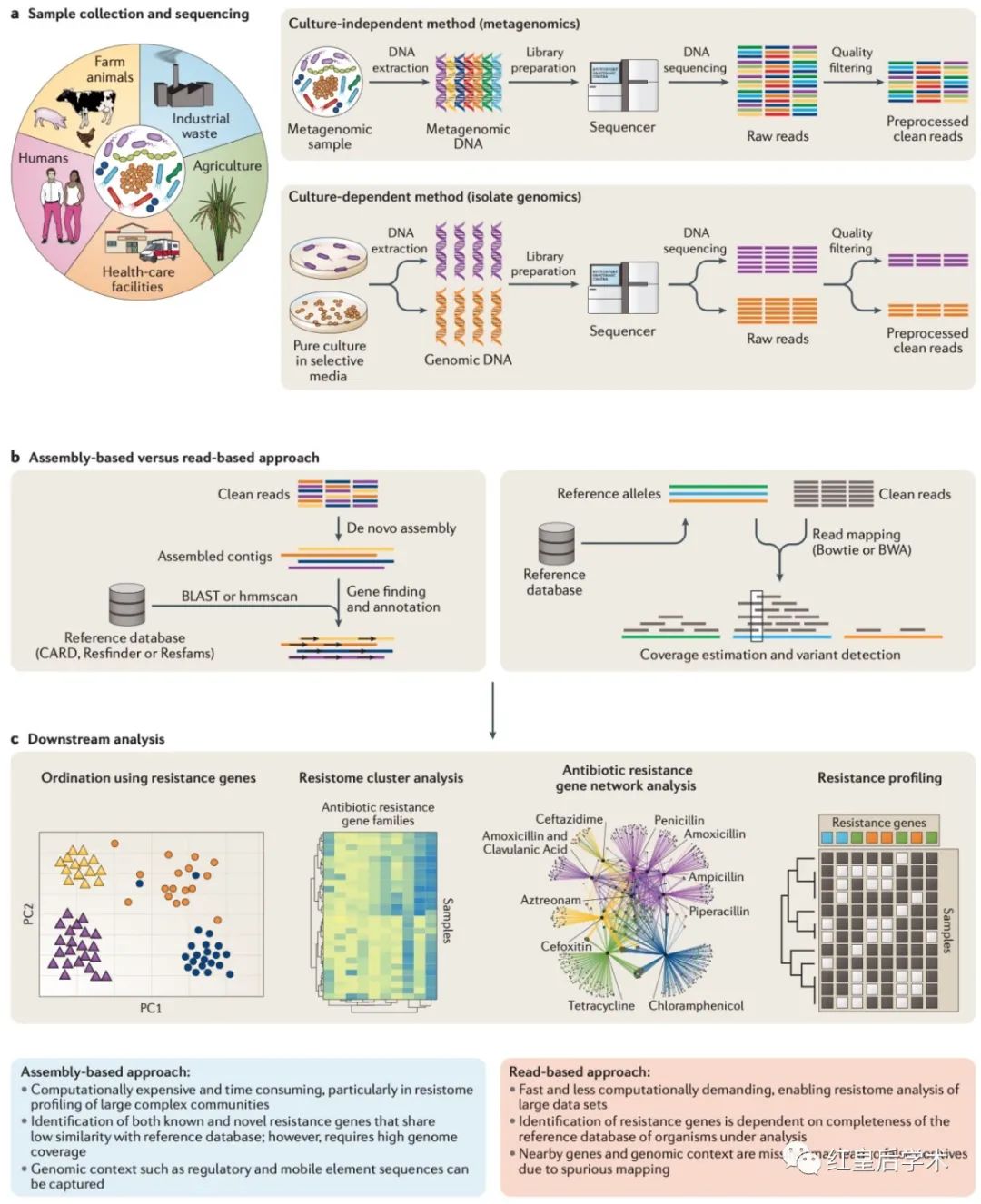
基于拼接的方法
从短读长数据进行基于全基因组测序的细菌基因组的从头组装,通常使用基于图论(De Bruijn graph,DBG)的工具,例如SPAdes、Velvet、ABySS和SOAPdenovo。
在这种方法中,测序读段被分为长度为k(其中k小于读取长度)的较短的重叠子序列(称为k-mers),并用于形成网络图,然后,拼接软件通过遍历每个网络的边找到一条最佳路径,从而重建基因组序列。
尽管DBG方法在处理大量测序数据方面计算效率很高,但是它受到测序过程中引入错误的极大影响,测序数据中的错误会导致假k-mers形成拼接碎片。一些拼接工具(例如SPAdes和Velvet)在找到最佳路径之前,会通过试探法消除这些错误。
组装宏基因组数据比单个细菌基因组的组装更为复杂,这是因为算法需要考虑具有未知系统遗传关系的不同生物体的未知丰度。在单基因组组装中,软件可以使用整个基因组的统一测序覆盖率来纠正测序错误并鉴定重复序列和质粒。但是宏基因组数据中不同生物的覆盖率不均匀,因此很难识别重复序列。不同物种中较长的一致序列是的分配短的测序读段到确定的细菌更加困难。
因此,为单基因组组装开发的算法不能直接应用于组装宏基因组,目前已经开发了几种专门用于宏基因组的拼接组成程序来克服这些困难,主要的方法是对不均匀的测序深度进行DBG的分区或优化,包括IDBA-UD、MEGAHIT、MetaSPAdes和MetaVelvet。
但是,目前没有哪个组装程序能脱颖而出,成为可以准确地重建已知基因组并捕获真实数据集中大多数分类多样性的最佳组装程序。生物学因素(例如样品来源和微生物群落结构)和技术因素(例如文库制备方法、测序深度和测序平台选择)都影响组装程序生成准确且较大的重叠群的能力。
因此,建议在一个样本子集上应用多个组装程序,以确定对于给定数据集的最佳组装结果。
组装完成之后,通过预测重叠群上的蛋白质编码区,然后使用基于相似性的搜索工具(例如BLAST、USEARCH或DIAMOND)将其与抗菌素耐药性参考数据库进行比较,为基因组或宏基因组重叠体标注耐药性遗传决定因子。
尽管测序数据与参考序列之间的成对比对是从重叠群中表征菌群的最常用方法,但数据库中天然的对人类相关生物的倾向性会影响输出的结果,因此选择适当的数据库进行拼接重叠群于参考序列的比对是非常重要的。
如果有足够的覆盖率,基于组装的方法可以构建完整的基因组或具有蛋白质编码基因、调控序列信息和完整的周围基因组背景的大型重叠群,这可以用于研究耐药性单元涉及的相关基因和生物学途径。
宏基因组数据的拼接和注释可以识别与参考数据库中已知序列差异更大且缺乏同源性的耐药基因, 然而,从头组装和注释的过程在计算上是昂贵、费时的,并且与基于参考数据库的组装或基于read的比对方法相比,需要更高的基因组覆盖率,这对于所有样品都是一个不小的困难,特别是那些微生物多样性高、分类组成不均的宏基因组样品。
基于read的方法
可以不经过基因组组装直接检测样本中的抗菌素耐药基因,主要的方法是通过使用成对比对工具(例如Bowtie2或BWA)将read于参考数据库比对,或者将read拆分为k-mer之后映射到参考数据库上。
SRST2是一种被广泛应用的工具,其利用Bowtie2将reads与自定义参考数据库进行比对,以预测样品中的抗菌素耐药基因。KmerResistance将read拆分为k-mers,对其进行参考数据库映射,以预测耐药基因和相关物种。
即使在存在污染物的情况下,例如,由于实验室或宿主污染而在原始读数中产生背景噪音,以及在没有足够的reads可用于从头组装的样品中,两种方法都可以识别抗菌素耐药基因,但这两种方法都无法预测由单核苷酸多态性导致的抗药性。
与之相比,ARIBA使用一种混合方法,数据库中的参考序列首先使用CD-HIT进行聚类形成独立的类群,之后对同一类群的参考序列进行独立组装,将得到的重叠群与最接近的参考序列进行比较以鉴定等位基因变体。此外,ARIBA还提供有关基因是否完整的信息,并报告序列变异及其潜在影响(例如错义、无义或移码突变以及小的插入和缺失)。
对参考序列进行聚类,并使用来自聚类的代表性序列与测序reads进行比对,可大大提高比对的准确定,但使用单个线性代表性基因序列掩盖了聚类内基因的亚型和亚家族之间微妙而重要的变异。
为了解决这种信息丢失的问题,GROOT作为一个新发布的宏基因组抗性组识别工具,构建了参考基因集的变异图谱,并将read与其比对,变异图谱是双向非循环序列图,表示一个种群内的的总体序列变异。reads与变异图谱的比对可以有效消除参考偏差,并有助于准确标记抗菌素耐药基因。
在将序列与变异图谱进行比对之前,通过Burrows-Wheeler变换或哈希图建立索引,可以显着提高大规模测序reads与变异图谱的的映射率。
基于read的方法通常快速且对计算的要求较低,因为它绕过了从头组装、蛋白质编码基因预测和与公共数据库的成对比对的过程。
由于这个原因,近年来,基于read的方法获得了广泛的关注,尤其是在临床诊断中,基于实时测序的耐药性预测至关重要。
选择正确的方法
目前,关于哪种序列分析方法更好尚无共识,分析的选择主要取决于测序的类型以及计算资源的可用性和研究目的。
两种方法都需要权衡,因为与直接基于reads的分析相比,基于拼接的分析会丢失一些信息,但可以识别蛋白质编码基因并用于上游和下游调节元件的研究,而直接基于reads的分析缺乏基因的位置信息。新的测序技术,例如长读测序和源自染色体构象捕获的方法,正在通过提高基因组组装的保真度来减少拼接过程中的信息丢失。
基于reads的方法可随着不断增加的测序数据和抗菌素耐药基因参考数据而不断的完善,更重要的是,它们能够从复杂群落中存在的低丰度生物体中鉴定出抗菌素耐药基因,由于组装不全或组装不良,基于拼接的方法可能会遗漏这些基因。
但是,将reads直接映射到大数据集可能会增加预测的假阳性,因为源自蛋白质编码序列的reads可能会由于局部序列同源性而虚假地与参考基因匹配,因此,参考数据库是否全面并且包含参考基因的所有变体非常重要。
当从大型和复杂的群落(例如土壤和海洋)中鉴定抗菌素耐药基因时,数据库的选择尤为重要,因为不完整的数据库可能会漏掉未被充分研究的,特征较少的环境群落中存在的抗菌素耐药基因。
相比之下,经过深入研究的样本类型,例如人的肠道,即使对于低丰度的微生物,基于reads的方法也可以得到可靠的结果。
由于缺乏参考序列,对于不同环境中抗菌素耐药基因的分析可能存在低估的现象,ShortBRED被开发出来用于解决该问题,该方法可以在宏基因组学数据集中快速、准确地分析抗性组。
ShortBRED首先从参考数据库中鉴定出代表抗菌素耐药蛋白家族的标记序列,之后将测序reads与这些标记序列进行比对计算相关抗菌素耐药蛋白家族的相对丰度。这宗方法已经成功的用于量化大型和复杂宏基因组学数据集(包括人类、动物和环境数据集)中抗性基因的丰度。
宏基因组样本中抗性组的下游分析可以类似于分类学和功能分析进行,具体信息可以参考This detailed review discusses the best strategies used in shotgun metagenomics studies。
Box2:基于测序的创新
染色体构象捕获技术
染色体构象捕获使用交联、连接和短读长测序来了解细胞内遗传物质的空间关系,这种方法已经有效地应用于细菌分离株和宏基因组样品中。利用来自染色体构象捕获的空间信息,研究人员可以增加对抗菌素耐药基因调控的了解,并阐明更复杂的耐药机制。
例如,这些数据可能有助于解决抗菌素耐药性基因与其上游基因之间的关系,因为基因空间共定位可能表明一些调控功能。染色体构象捕获也可用于从染色体基因中鉴定质粒基因,由于质粒在水平基因转移中的作用,这一点对于抗菌素耐药性非常重要。
最吸引人的是染色体构象捕获对宏基因组样本的适用性,宏基因组样本中的交联信息可以大大降低了宏基因组组装的复杂性,它使研究人员能够按细胞对序列进行分组,因此,结合染色体构象捕获信息的基于拼接的的抗菌素耐药性注释方法可能能为可靠。
长读长测序
通过单分子实时测序或纳米孔测序进行的长读测序可产生10kb至100kb以上的序列,长的序列在研究纯培养细菌和宏基因组学样品中的抗菌素耐药性方面具有很多优势。
长读测序可大大降低纯培养细菌和宏基因组学样品序列组装的复杂性,在某些情况下,可提供完整的细菌基因组(比短读错误率更高,这意味着有时需要短读长和长读长测序相结合)。更好的组装可以对基因组背景进行更深入的解释,提供与染色体捕获类似的好处。
与染色体捕获类似,长读长测序可以更轻松地解析质粒序列,从而可以进行更深入的研究来了解抗菌素耐药基因的水平基因转移。
此外,这两种长读长测序方法都提供了有关DNA甲基化的信息,这可以租住宏基因组样品中基因组的组装。
最后,长读长测序运行时间往往比短读测序快,这对于患者预后的潜在临床部署很有用,由于其实时测序的方式,纳米孔测序在这一领域尤其出色,这些实时数据已被用于快速的预测肺炎链球菌中由AST无法获取的未知抗菌素耐药基因的识别,只需要几分钟就可以得到结果。
转录组
RNA测序等转录组学技术有助于细菌基因表达的分析,这些表达数据具有填补抗菌素耐药性知识空白的潜力。转录数据可以将基因型抗菌素耐药性数据与表型抗菌素耐药性结果联系起来。
在存在抗菌素耐药基因但未发现耐药性表型的情况下,或在无明显抗菌素耐药基因但已确认耐药性的情况下,转录组数据可弥补基因组数据的不足,并有助于发现新型耐药基因。
转录组学还具有鉴定导致抗菌素耐药性的组合基因效应的潜力,此外,转录组学可以成功地鉴定非编码调控RNA导致耐药细菌表型的情况。
抗菌素抗性基因数据库
无论是基于拼接和基于read的方法,预测抗菌素耐药基因在很大程度上都取决于经过整理的抗菌素耐药性基因数据库,这些数据库将已知的耐药性遗传决定因素与它们赋予细菌的耐药性表型联系起来,这些数据库通常代表从多项研究中收集的信息,其中包括携带特定抗菌素耐药基因的细菌的AST实验。
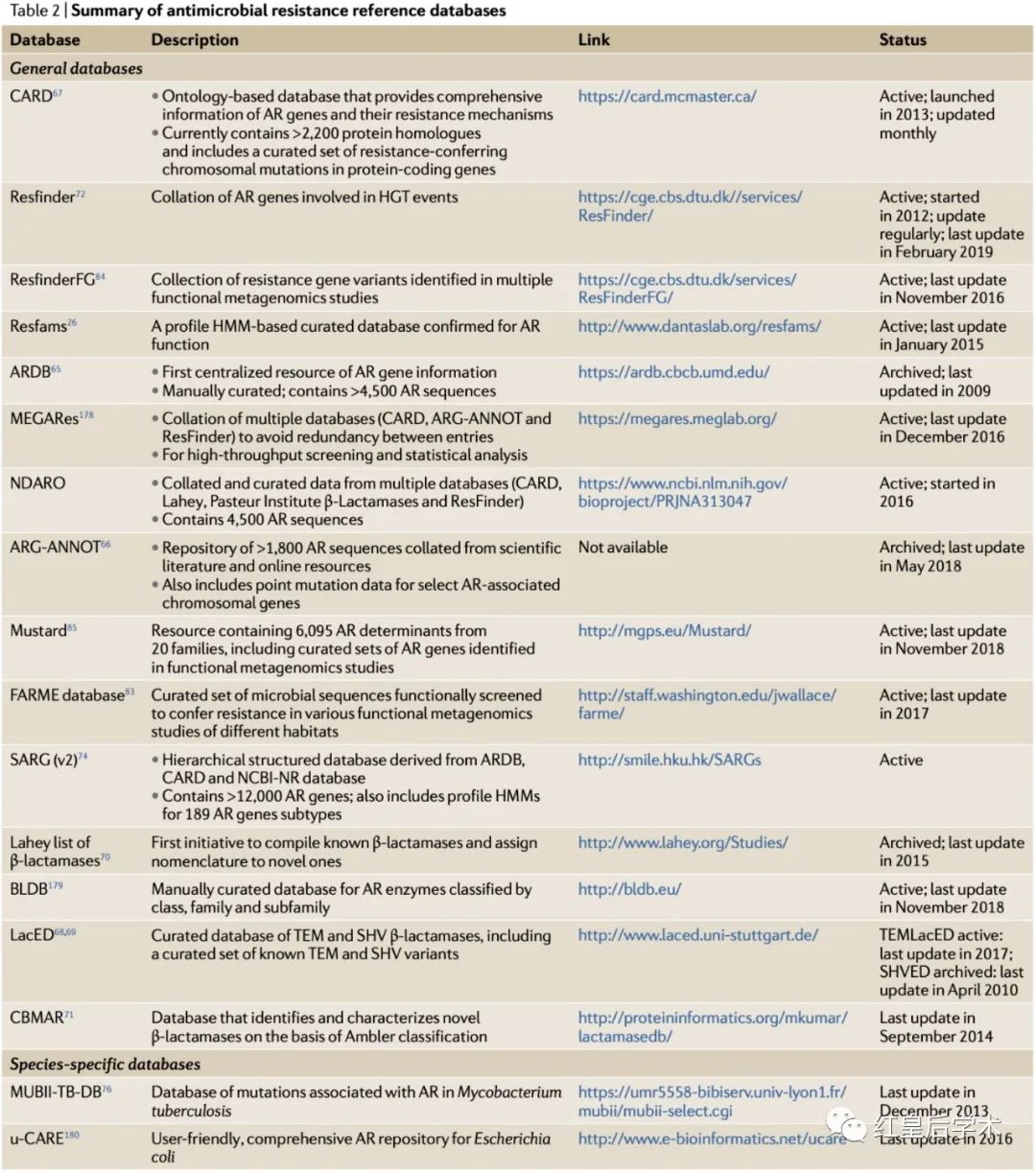
通用/特定的数据库
公共数据库在其所涵盖的抗性机制的范围以及它们提供注释的信息类型方面很大差异。
通用的抗菌素耐药基因数据库,例如已经不再更新的ARDB活依然更新的ARG-ANNOT和CARD,涵盖了广泛的抗菌素耐药基因及其机理信息。
而特定的抗菌素耐药性数据库则提供了有关特定基因家族或物种的全面信息,例如LacED、Lahey、NCBI β-Lactamase Alleles Initative以及CBMAR,这些数据库只关注β-内酰胺酶,这是一种抗微生物酶,可促进β-内酰胺抗菌素中关键β-内酰胺环的水解,从而使细菌免受抗菌活性的影响。
Resfinder是一个基于Web的独立工具,与其他需要重叠群作为输入的数据库不同,Resfinder还接受短的reads为输入用于检测已测序或部分测序的细菌分离株中含有的抗菌素耐药基因。
2017年,Resfinder更新了其基于Web的服务工具,其可以通过PointFinder识别染色体体突变,但是,只有少数有限的病原微生物(弯曲杆菌、大肠杆菌、结核分枝杆菌、淋病奈瑟氏球菌、恶性疟原虫和沙门氏菌)可鉴定出具有耐药性的染色体突变。
与Resfinder相似,CARD同样提供了自己的分析工具,称为RGI,它使用经过整理的抗菌素耐药基因检测模型来预测内在的抗菌素耐药基因、专用抗菌素耐药基因和药物靶点突变后获得的耐药基因。RGI使用两种检测模型:用于检测抗菌素耐药蛋白功能同源物的Protein Homologue Model和用于检测赋予耐药性的突变的Protein Variant Model。
ARGs-OAP使用由ARDB和CARD构建的自定义数据库SARG,以及一种混合了UBLAST和BLASTX的算法,展示了全面的数据库在基于宏基因组序列抗菌素耐药基因注释中的重要性。
存在针对病原菌或模式细菌的特定物种数据库,例如Tuberculosis Drug Resistance Database和MUBII-TB-DB,这些物种特定的数据库对于了解这些特定生物体的耐药性具有非常的作用,但同时也强调了考虑将抗生素耐药性基因纳入其系统发育研究的重要性,特别是对那些含有先天性耐药性的细菌。
以物种为中心的数据库可以快速有效地处理新的抗菌素耐药基因和染色体突变,并可以提供快速的初步筛选以鉴定其特性。事实证明,这种筛查对结核分枝杆菌等病原体非常有效,在这些病原体中,水平基因转移事件很少见,耐药性主要来自染色体突变。
CRyPTIC Consortium和100,000 Genomes Project证明了物种特异性数据库的有效性,针对结核分期杆菌的数据库对四种一线抗结核药物的耐药性预测均具有90%以上的敏感性和特异性。
尽管这些工具正在朝着正确的方向前进,但仍需要一个持续更新的全面数据库,该数据库应具有广泛的基因元数据,并且必须同时具备能够发现点突变和远距离同源性的能力。
基于隐马尔可夫模型的数据库
上述数据库的主要局限性在于它们所包含的抗菌素耐药基因严重偏向人类病原体和易于培养的模式生物,从而很难从未被培养的细菌中鉴定新型的或远距离同源的耐药基因。
这种偏好性使得在不常见细菌中鉴定抗菌素耐药基因更为困难,克服这种偏好性的一种潜在解决方案是使用隐马尔可夫模型(HMM)数据库,从已知序列的多序列比对得出,HMM可以找到具有相似功能但序列同一性较低的序列。
Resfams是抗菌素耐药蛋白的HMM数据库,该数据库通过来自CARD、LacED和Lahey数据库的代表性抗菌素耐药蛋白序列的手动多序列比对进行构建。
Resfams数据库的作者声明,与依靠基于BLAST的方法进行基因鉴定的其他数据库(如ARDB和CARD)相比,Resfams可以识别出数量更多的新型抗菌素耐药基因和已知抗菌素耐药基因的远距离同源物。
对手动选择的抗菌素耐药基因集的直接比较显示,与基于BLAST的CARD和ARDB搜索相比,Resfams在土壤和人类肠道菌群中鉴定出的抗菌素耐药基因的数量提升了64%,这种提高的敏感性证明了HMM在非临床样品序列注释的可行性。
但是,基于HMM的方法可能具有较差的特异性,会产生较高数量的假阳性结果,并且可能无法区分功能密切相关的蛋白质家族。
目前,Resfams包含166个代表主要抗菌素耐药基因家族的HMM,与基于BLAST的数据库相比,基于HMM的抗菌素耐药性数据库在鉴定未充分研究的环境样品中大量多样的耐药性决定因素方面可能具有重要价值。
但是,当前基于HMM的数据库无法识别由染色体突变引起的耐药性,为了进一步促进在大型复杂环境中检测抗菌素耐药基因,建立了FARME数据库,其包含不属于目前已有数据库但是通过功能宏基因组研究发现具有耐药性的微生物序列。除了预测的蛋白质编码的抗菌素耐药序列外,FARME数据库还包括调控元件、移动遗传元件和位于抗菌素耐药基因周围的预测蛋白质。
类似的,通过汇总针对23种抗微生物药物选择的功能宏基因组学数据,建立了ResfinderFG数据库,当将该数据库与Resfinder数据库进行比较时,发现同一个基因出现了不同的耐药行结果,这种结果可能代表了将假定的抗性决定簇克隆到大肠杆菌中时与在其天然细菌宿主中表达时相比,结果可能存在一定的差异。
Mustard数据库使用一种整合3D蛋白结构的创新方法来帮助预测耐药性基因,当这种方法应用于宏基因组样本时,它预测了超过6,000个抗性基因,而同样的数据使用BLASTP只鉴定的67个基因和使用Resfinder只鉴定的50个基因,说明Mustard的方法具有更高的敏感性。
存在的挑战
虽然已经取得了长足的进步,但是抗菌素耐药基因的鉴定和研究依然面临着成本和分析速度的限制,一个主要的瓶颈是缺乏有效的管理策略。除少数例外,抗菌素耐药性数据库都缺乏有效且可持续的更新流程。
大多许抗菌素耐药基因都是根据其核酸或蛋白质序列的相似性来获得匹配的名称,导致了命名规则的冲突,冲突的基因名称和同义词会在数据库之间造成冗余,并给用户带来一些误解。例如在某些数据库中,二氢叶酸还原酶被称为dhfr,而在其他数据库中被称为dfrA。
通过序列的一致性分配基因名称会加剧这种情况,存在许多不同的基于序列一致性的系统,用于将基因名称分配给新的抗性基因。这些系统提供了不同的临界值,并且参考文献也并不一致。
抗菌素耐药性基因数据是一个不断扩展的数据源,水平基因转移事件和选择压力带来抗菌素耐药性突变,需要采取积极的生物固化策略,从而可以在识别输入后对其进行整合。
mcr-1基因于2016年首次在中国人的一个细菌分离株重被发现,随后在世界各地均有检出,这种新型抗菌素耐药基因的增殖过程表明需要经常更新并规划数据库。如果这些操作能被正确的实施,它们有助于快速收集最近发现的耐药性决定因素的流行病学数据。
由于所有下游分析都依赖于参考数据库的准确性,因此抗菌素耐药基因的注释注释应是一项持续的工作,建立最佳的数据整合流程,为新发现的基因系统地分配注释,防止误解,将为公共卫生和基础科学带来不可估量的好处。
当前的抗菌素耐药性数据库的另一个重要局限性在于它们专注于蛋白质编码耐药性基因的鉴定和表征,而忽略了其他潜在的耐药机制,例如基因组变化或核糖体RNA基因从头突变、调控元件和药物靶点突变等等,CARD和Resfinder正在试图解决这个问题。
功能宏基因组
除基于序列的宏基因组学外,功能宏基因组学是一种强大的、不依赖于培养技术的、无序列偏差的抗性组鉴定方法。
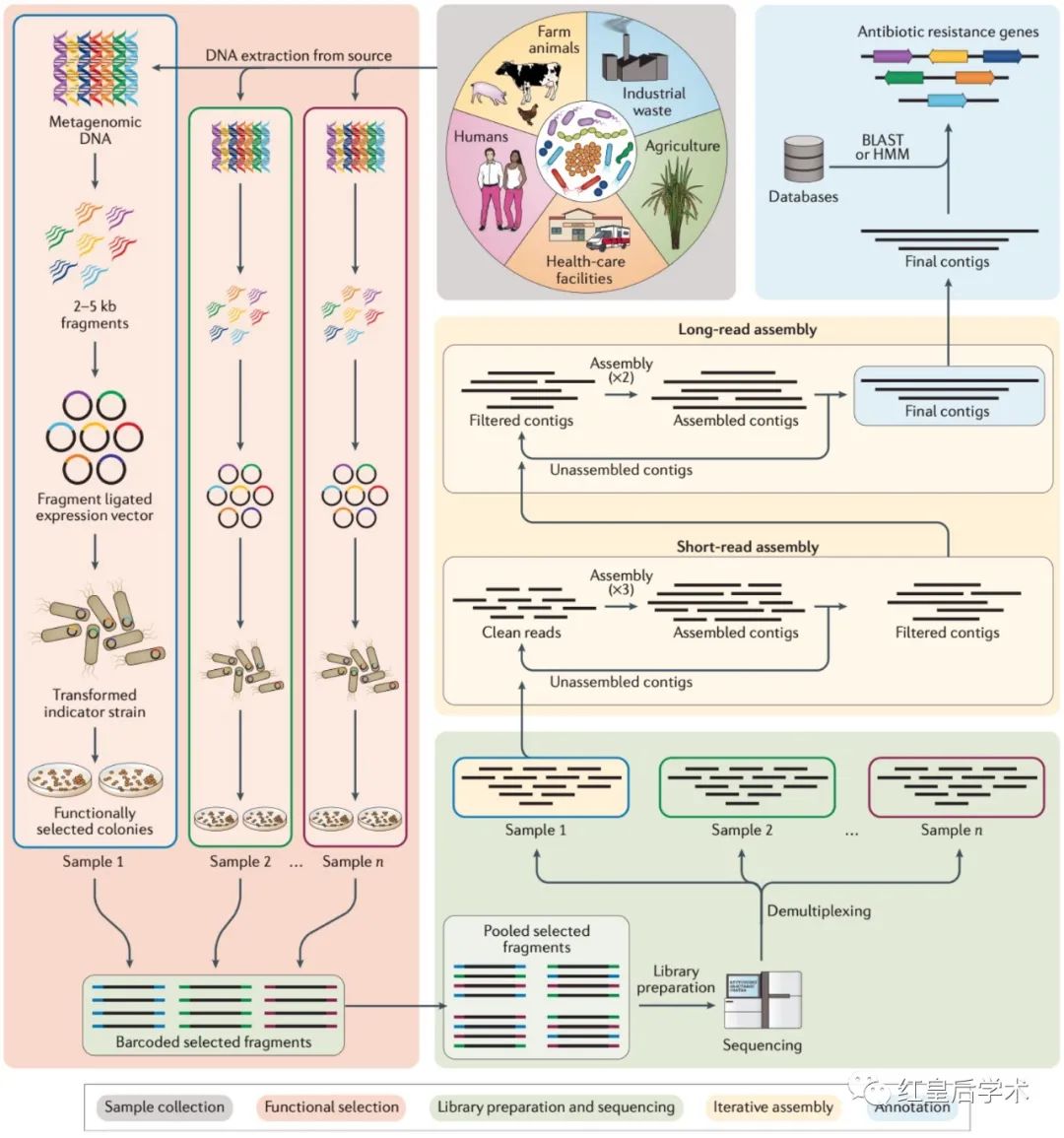
在这种方法中,通过将从样品中提取的总群落DNA克隆到表达载体中来生成宏基因组文库,该文库被转移进入一种抗菌素易感的指示宿主菌株中,并通过对野生型宿主具有致死性的选择性培养基上进行铺板来测定抗菌素耐药性,最后对存活的细胞中的插入片段进行测序,对所得序列进行组装和注释。
PARFuMS是一个定制的计算流程,它使用Velvet和Phrap将功能宏基因组的数据拼接成contigs,并使用MetaGeneMark和Resfams注释抗菌素耐药基因。
这种方法可以对大基因组含量的数据进行高通量分析,而且耐药性表型可以直接与基因相关联,从而无需培养单个的耐药基因载体。
功能宏基因组学使人们能够发现新的抗菌素耐药机制及其相关基因,一个这样的例子是最近发现的四环素破坏酶对四环素的抗性机制,使用土壤功能宏基因组学发现了九种通过酶促失活赋予四环素抗性的基因,进一步的分析和生化特性表明,这些酶以FAD依赖的方式催化四环素氧化,从而使四环素失活。
尽管先前的研究显示了功能宏基因组学的优势和实用性,但是这种方法同样有一定的局限性。例如,基因必须在其天然微生物宿主之外具有功能,才能通过功能宏基因组学选择进行鉴定。
很多时候,重组表达宿主(如大肠杆菌)与原始宿主(例如某些革兰氏阳性生物)之间的差异不会赋予同一基因相同的表型,因此,需要系统发育上多样化的宿主群体。
另外,在基因组范围之外的基因,例如协同调控元件,在重组表达宿主中可能具有与原始宿主不同的表型,因此,重要的是通过功能宏基因组学筛选鉴定出的新型抗菌素耐药基因必须在生物学和生物化学上进行表征,当前功能宏基因组学方法的扩展和发现新抗性基因的新技术的开发是值得研究的方向。
机器学习用于耐药基因预测
已有众多研究探索了可以用于研究抗菌素耐药性的机器学习算法,重点强调了其在直接根据基因型预测耐药性表型中的作用。
机器学习方法可以分为为监督学习方法或无监督学习方法。
在监督学习中,可以利用具有感兴趣结果的训练数据集来构建预测模型,该模型可以进一步应用于查询序列以预测其结果。
多项研究使用基因存在与否以及AST结果作为特征来创建模型训练集,在一项研究中,使用logistic回归方法开发了基于14个基因参数和3个分子分型标记的模型,该模型可以使用公开可用的基因组数据和患者分离菌株识别万古霉素敏感的金黄色葡萄球菌。通过leave-one-out验证方法测试了该模型性能,结果表明分类精度为84%,尽管此准确度水平不符合临床标准,但该方法提供了重要的概念证明,有助于开发更复杂的模型来鉴定抗菌素耐药性。
另一项研究比较基于规则和基于机器学习的方法对抗菌素耐药性的预测,并显示基于机器学习的方法具有更高的准确率。
最近的研究和工具使用衍生自具有耐药性和抗菌素敏感的物种的全基因组的k-mers及其AST结果来开发预测模型。
Mykrobe是一种快速的k-mer筛选工具,用于鉴定金黄色葡萄球菌和结核分枝杆菌中的抗菌素耐药基因和SNP,它利用相同物种抗性和易感等位基因的精细化遗传信息来构建这两个类别的DBG,并将从测序reads中获得的k-mers映射到这些DBG上,Mykrobe预测因子在独立的验证集上分别显示出对金黄色葡萄球菌和结核分枝杆菌预测的敏感性分别为99.1%和82.6%,特异性分别为99.6%和98.5%。
RAST是另一种基于k-mer的工具,其使用基于PATRIC数据库的机器学习分类器(AdaBoost)来识别特定病原体中目标特异性的抗菌素耐药基因基因。RAST在从每个基因组重叠群衍生的k-mer数据上进行训练,将这些k-mer计数转换为1s和0s的二进制矩阵,以描述该基因组中是否存在特定的k-mer,然后将二元矩阵与AST结果一起用于形成分类模型,并确定与抗性相关的推定k-聚体。RAST分类器可以鉴定鲍曼不动杆菌中的碳青霉烯耐药性、金黄色葡萄球菌的甲氧西林耐药性以及肺炎链球菌的β-lactam和抗曲莫唑耐药率,准确度为88–99%。
所有机器学习分类器的一个主要缺点是其对训练数据或现有知识库的依赖,为了将机器学习分类器应用到临床诊断中,需要大量包含正确的基因型数据和相关AST数据相关的数据集,以建立一个有效而强大的基于机器学习分类器来识别细菌的耐药行。
除了区分抗药性微生物和对药敏性微生物外,目前还采用机器学习方法来预测宏基因组学数据中的抗菌素耐药基因。
DeepArgs是一种新近建立的工具,可通过深度学习来识别抗菌素耐药基因,根据CARD和ARDB的精选数据集以及Uniprot蛋白数据,DeepArgs建立了抗菌素耐药蛋白和非抗菌素耐药蛋白之间的差异矩阵,并用于训练两个深度学习模型:用于分析拼接结果的DeepArg-LS和用于分析reads的DeepArg-SS,这些模型可用于预测新测试数据中的抗菌素耐药基因。
尽管将机器学习应用于抗菌素耐药性预测和分类是有前途的,但在将这些技术用于快速诊断并取代传统的培养技术和AST方面,还有很长的路要走。
结论和展望
抗菌素耐药性是主要的公共卫生威胁,监测和了解抗菌素耐药性的流行、机理和传播是个体患者护理和总体感染控制策略的重点。
尽管已经取得了长足进步,但对抗菌素耐药性检测和理解的障碍仍然存在,测序和自动化的抗菌素耐药性检测仪器的成本正在降低,但是启动和运营成本仍超过许多医疗保健的预算,这些技术进一步的降低成本对于其大范围应用至关重要。
准确确定耐药性决定因素以及抗菌素耐药性基因谱与抗菌素治疗结果的相关性,将有助于制定个性化的治疗方案,这种方法的成功在很大程度上取决于公开的抗菌素耐药基因数据库的全面性和质量,这些数据库在生物测定和计算工具的开发中起着重要作用,从而扩大了我们在单个菌株和微生物群落中检测耐药基因的能力。
尽管在建立全面的抗菌素耐药性基因数据库方面已取得进展,但不同数据库缺乏统一的标准和较长的更新间隔阻碍了它们的应用潜力。
而且,复杂的耐药机制很难在抗菌素耐药性数据库中被捕获,例如,耐药性可能源于多个基因之间的上下游关系,例如在碳青霉烯耐药性可能是由于广谱β-内酰胺酶和外排泵或孔蛋白不渗透性的结合而引起的。抗性甚至可能通过正常基因的过度表达而发生,例如编码外排泵的基因,而检测这些抗性机制需要转录水平的定量数据。
这些复杂的耐药机制与已知的抗菌素耐药基因可能不总是表达的事实相结合,导致难以从基因型抗菌素耐药性数据中准确预测耐药性的表型。
机器学习算法在预测耐药性表型方面取得了进展,但是这些技术倾向于针对特定的细菌,并且对于一般临床部署而言还不够准确。
为了实现根据基因型数据进行表型预测的目标,我们需要更全面的数据库,这些数据库将特定的抗菌素耐药基因与特定的AST结果相关联。更重要的是,这些数据库应包括具有完整序列的细菌、抗菌素耐药基因基因预测的元数据以及有确切抑菌区域大小或最小抑菌浓度的AST结果,而不是仅仅是分类准则,AST和基于序列的耐药基因预测的并行改进将加减轻耐药性性的临床影响。
尽管存在一些可以进行新型抗菌素耐药基因鉴定的技术,例如功能宏基因组学,但这些技术对于鉴定抗菌素耐药基因的类型仍然存在巨大的缺陷,迫切需要确定新型抗菌素耐药基因机制的创新方法。
此外,需要有稳定的模型来预测哪些抗性基因会在医疗机构内的本地水平以及国家之间的全球范围内传播,这些模型可能不仅需要结合抗菌素耐药基因序列和机理,而且还需要结合基因组背景、宿主细菌种类和地理位置。
快速、准确地鉴定分离细菌的和宏基因组样品中的耐药基因将增强临床医生制定细菌感染治疗计划的能力,从而为将来使用常规的基于序列的个性化医学提供便利。这也将简化抗菌素耐药性的监测工作,并使资源贫乏地区能够从测序成本的快速下降中更加充分地受益。
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
文献阅读 热心肠 SemanticScholar Geenmedical
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA

为开发者提供学习成长、分享交流、生态实践、资源工具等服务,帮助开发者快速成长。
更多推荐



 4
4 0
0- 0



 已为社区贡献15条内容
已为社区贡献15条内容

所有评论(0)