生信分析-本地BLAST
一. 本地blast简介本地Blast(Basic Local Alignment Search Tool),是基于本地的比对搜索工具,可以在自己建立的数据库进行blast搜索,与NCBI的在线blast相比,其速度快,搜索范围更小,且在没有互联网的情况下可以进行,比如已知道大麦的一个基因,并已经明确其功能,现在要在大麦中找序列相似度高的基因,就可以在本地建库,即建立大麦的数据库,然后blast找
一. 本地blast简介
本地Blast(Basic Local Alignment Search Tool),是基于本地的比对搜索工具,可以在自己建立的数据库进行blast搜索,与NCBI的在线blast相比,其速度快,搜索范围更小,且在没有互联网的情况下可以进行,比如已知道大麦的一个基因,并已经明确其功能,现在要在大麦中找序列相似度高的基因,就可以在本地建库,即建立大麦的数据库,然后blast找同源序列。
二. 本地blast的安装
1.NCBI下载本地版blast
[下载链接]:https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/下载与自己电脑系统相适应的版本,这里我下载ncbi-blast-2.11.0

2.安装本地blast程序
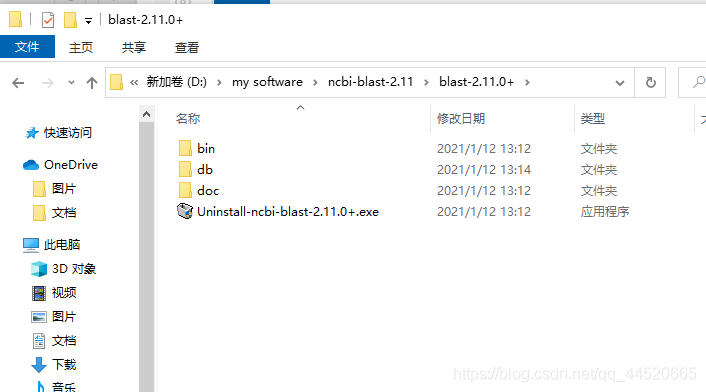
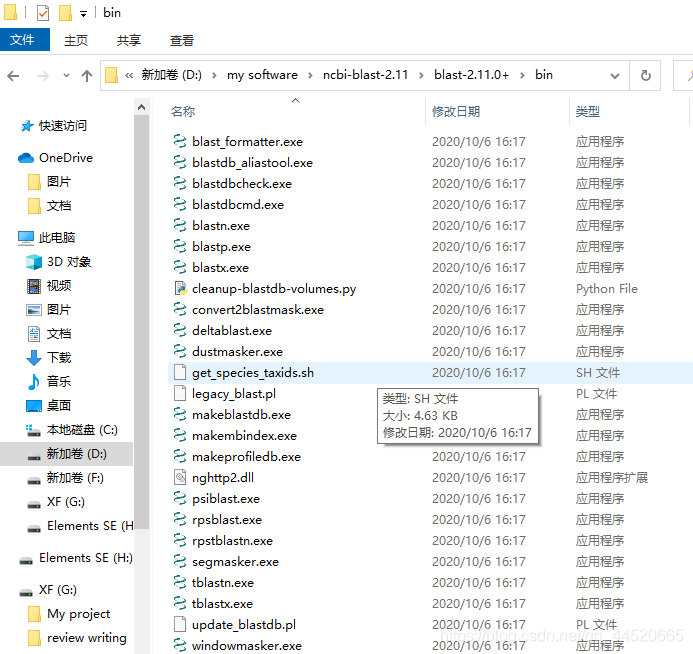
下载后双击安装,可通过比较程序大小判断是否下载完全,生成bin和doc两个子目录,其中其中bin是程序目录,doc是文档目录,也可点开查看程序的完整性,然后在目录下新建文件夹,重命名为db
3.用户环境变量设置
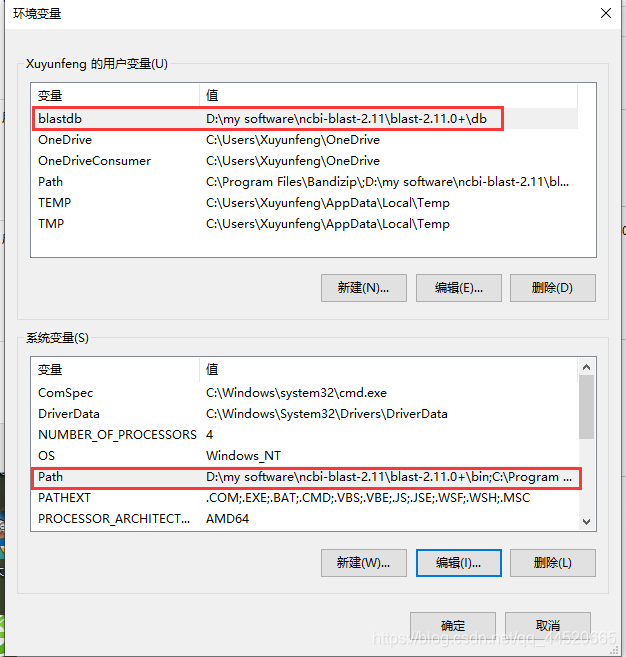
此电脑-属性-高级系统设置-环境变量
在用户变量下方:新建-变量名:balstdb,变量值为电脑安装好新建的db文件夹的路径
在系统变量下方:Path-添加变量值为电脑上bin文件夹位置
三.建立本地blast数据库
1.数据库下载
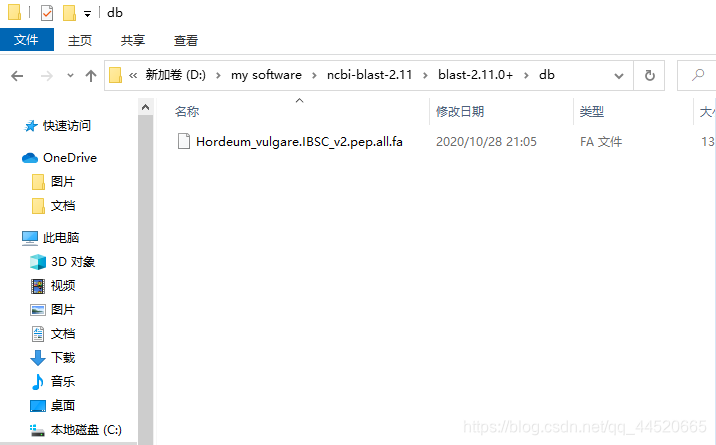
ensemble等数据库下载大麦的pep数据,并将其解压到db文件夹中
2.数据库格式化
在db文件夹中未选中任何目标的情况下,按住Shift,在文件夹空白处右击鼠标,选择在此处打开PowerShell窗口,打开如图所示窗口。我用的是win10的系统可以通过这种方式快速进入db文件夹的位置,如果是其他版本的windows系统,可能需要通过运行cmd进入db文件夹所在的位置。
attention:文件在当前Windows powershell所打开文件夹下因此不需要代入路径
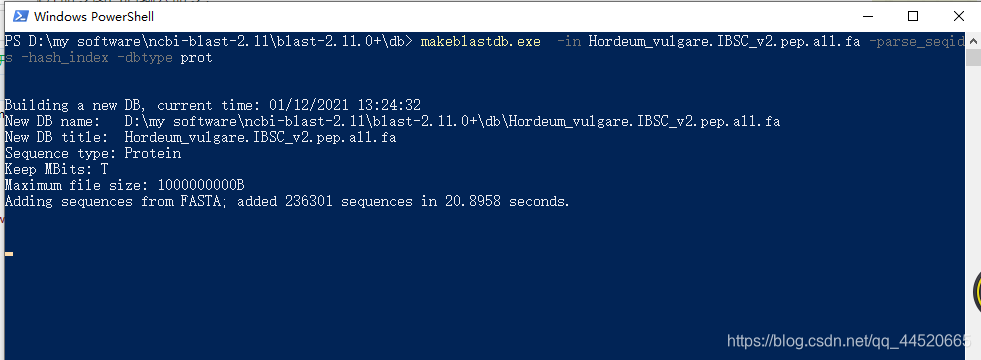
运行命令:
makeblastdb.exe -in Hordeum_vulgare.IBSC_v2.pep.all.fa -parse_seqids -hash_index -dbtype prot
其中Hordeum_vulgare.IBSC_v2.pep.all.fa 为你要格式化的数据库的名称,根据你自己的数据库的名称进行改动,记得加上后缀名.fa;dbtype后的prot表示数据库的类型,prot表示氨基酸序列的数据库,如果是核苷酸序列则用nucl。命令行中数据库格式化完成后显示下图
四.本地blast运行
1.query序列的准备
在D:\my software\ncbi-blast-2.11\blast-2.11.0+\db文件夹下创建target.seq.txt的文本文件,将需要查询到的序列以fasta格式保存到in.txt中,我们已1条barley的蛋白序列为例

2.query序列的查询
在D:\my software\ncbi-blast-2.11\blast-2.11.0+\db 文件夹下创建新建文本文件.txt的文本文件,使用blast的命令:
blastp.exe -task blastp -query target.seq.txt -db Hordeum_vulgare.IBSC_v2.pep.all.fa -out out.txt -evalue 1e-10 -outfmt 6 -num_threads 2
相关参数说明:
blastp.exe 程序执行命令,exe 前的程序根据自己的需要而换,包括blastn,blatp,tblastx等bin文件夹中所包含的程序;
-task 后面选择你所要用的程序,blastn,blastp,tblastx 等;
-query 后接查询序列的文件名称;
-db 后接格式化好的数据库名称;
-out 后接要输出的文件名称及格式,格式形式包括0-10,其中6和0最常用,可以自己尝试。
-num_threads 的参数设置可以根据自己电脑的性能进行设置,笔记本推荐不超过2,从而提高比对效率

保存后再将新建文本文件.txt重命名为Hordeum_vulgare.pep-blast.cmd,此时已经将一个文本文件修改为windows中的cmd命令,双击即可运行
blast结果说明
 每一列分别表示:
每一列分别表示:
A:Query_id
B:Subject_id
C:Identity
D:Align_length
E:Miss_match
F:Gap
G:Query_start
H:Query_end
I:Subject_start
J:Subject_end
K:E_value
L:Score
E值(Expect):表示随机匹配的可能性,例如,E=1,表示在目前大小的数据库中,完全由机会搜到对象数的平均值为1.E值越大,随机匹配的可能性也越大。E值接近零或为零时,具本上就是完全匹配了。通常来讲,我们认为E值小于10-5 就是比较可性的S值结果。我们可以想象,相同的数据库,E=0.001时如果有1000条都有机会S值比现在这个要高的话,那么不E设置为10-6时可能就会只得到一条结果,就是S值最可靠的那个。但是E值也不是万能的。它在以下几个情况下有局限性:
1)当目标序列过小时,E值会偏大,因为无法得到较高的S值。
2)当两序列同源性虽然高,但有较大的gap(空隙)时,S值会下降。这个时候gap scores就非常有用。
3)有些序列的非功能区有较低的随机性时,可能会造成两序列较高的同源性。
E值总结:E值适合于有一定长度,而且复杂度不能太低的序列。当E值小于10-5时,表明两序列有较高的同源性,而不是因为计算错误。当E值小于10-6时,表时两序列的同源性非常高,几乎没有必要再做确认。
一致性(Identities):或相似性。匹配上的碱基数占总序列长的百分数。
Score:得分值越高说明同源性越好;Expect期望值越小比对结果越好,说明因某些原因而引起的误差越小;Identities是同源性(相似性),例中所示比对的1299个碱基中只有35个不配,其他97%相同;
Gaps:是指多出或少的碱基或缺失的碱基数;缺失或插入(Gaps):插入或缺失。用"—"来表示。
此外比对的Strand则通 s. Start:和s. End判断,如上述结果的第三行. Star值大于s. End,则表示负链。
总结及补充:另外的方法
进行本地blast序列对比我们需要建立一个库文件(在线blast的库使用的是各大生物数据库的文件),输入命令:makeblastdb建库命令。
makeblastdb -in b.fasta -dbtype nucl -out b.fasta.blastdb
makeblastdb -in b.fasta -dbtype nucl -out b.fasta.blastdb[文件在当前Windows powershell所打开文件夹下因此不需要代入路径]
==注意空格不能少=
-in 后面是建库文件,我们使用较大的文件建库
-out 后面跟输出的库文件名,一般在第一步建库之后会生成三个文件nhr/nin/nsq这三个共同作为一个库进入下一步。
如果跑的是核酸序列,使用blastn
blastn -query a.fasta -db b.fasta.blastdb -out b.blast -outfmt 6 -evalue 1e-5 -num_threads 2
blastn -query a.fasta -db b.fasta.blastdb -out b.blast -outfmt 6 -evalue 1e-5 -num_threads 2
-query: 输入文件路径及文件名
-out:输出文件路径及文件名
-db:格式化了的数据库路径及数据库名
-outfmt:输出文件格式,总共有12种格式,6是tabular格式对应BLAST的m8格式
-evalue:设置输出结果的e-value值
-num_descriptions:tabular格式输出结果的条数
-num_threads:线程数[笔记本一般不超过2]
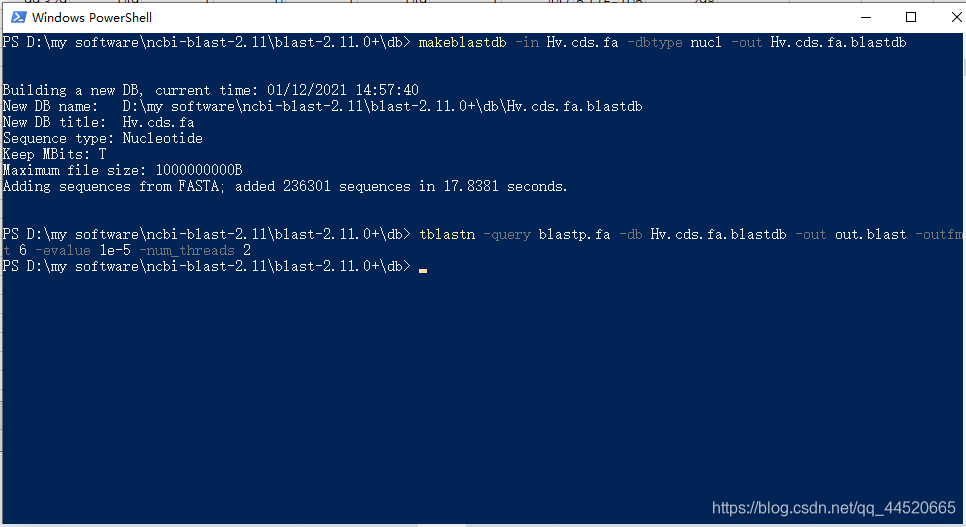
该图为我用蛋白质序列去核酸数据库跑tblastn所运行的命令
最后,附上“参考文献”
参考文章链接:
https://blog.csdn.net/zxpuls123/article/details/81407277
https://blog.csdn.net/qq_43337286/article/details/103120003

为开发者提供学习成长、分享交流、生态实践、资源工具等服务,帮助开发者快速成长。
更多推荐



 20
20 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)